
Subtractive Proteomics and Reverse-Vaccinology Approaches for Novel Drug Target Identification and Chimeric Vaccine Development against Bartonella henselae Strain Houston-1

 ,
,  , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pathogen Proteome Retrieval and Exclusion of Repetitive Sequences
2.2. Identification of Non-Homologous Proteins
2.3. Identification of Vital Proteins
2.4. Evaluation of Unique Metabolic Pathways
2.5. Subcellular Localization Analysis
2.6. Evaluation of Druggability in Essential and Unique Proteins
2.7. Screening of Gut Microbiota Protein
2.8. Prediction of Antigenic Membrane Protein
2.9. Protein–Protein Interaction
2.10. Prediction of T-Cell MHC-I Epitope
2.11. Analysis of Class I Immunogenicity, Antigenicity, Allergenicity, and Toxicity
2.12. Prediction of T-Cell MHC-II Epitopes
2.13. MHC-Restricted Alleles Clustering
2.14. Prediction of B-Cell Epitopes
2.15. Design of the Multi-Epitope Vaccine Construct
2.16. Antigenicity, Allergenicity, and Solubility Evaluation of the Designed Vaccine Construct
2.17. Secondary and Tertiary Structure Predictions, Refinement, and Validation of the Designed Vaccine Construct
2.18. Physiochemical Properties of the Designed Vaccine Construct
2.19. Disulfide Engineering of the Designed Vaccine Constructs
2.20. Molecular Docking of the Designed Vaccine Construct with Human Toll-Like Receptor 4 (TLR4)
2.21. Molecular Dynamics Simulation
2.22. Discontinuous B-Cell Epitope Prediction
2.23. Simulation of Immunity
2.24. Codon Optimization of the Designed Multi-Epitope Vaccine Construct and Its Virtual Cloning
2.25. Prediction of the mRNA Structure Encoding the Multi-Epitope Vaccine Construct
3. Results and Discussion
3.1. Pathogen Proteome RETRIEVAL, filtration, and Non-Host Homolog Protein Identification
3.2. Identification of Essential Proteins, Unique Metabolic Pathways, and Subcellular Localization
3.3. Assessing Druggability, Virulency, and Screening of Gut Microbiota Proteins
3.4. Prediction of Antigenic Membrane Protein and Its Interactions with other Proteins
3.5. Prediction of MHC-I Epitopes, Class I Immunogenicity, Antigenicity, and Non-Toxicity Analysis for Designing the Multi-Epitope Vaccine Construct
3.6. Prediction of MHC-II Epitopes for Designing the Multi-Epitope Vaccine Construct
3.7. Assessment of MHC Restriction and Cluster Analysis
3.8. Identification of B-Cell Epitopes for Designing the Multi-Epitope Vaccine Construct
3.9. Formulation of the Epitope-Based Subunit Vaccine
3.10. Allergenicity, Solubility, Antigenicity, and Physiochemical Features of the Designed Multi-Epitope Vaccine Construct
3.11. Secondary Structure Prediction of the Designed Multi-Epitope Vaccine Construct
3.12. In Silico Tertiary Structure Prediction, Its Refinement, and Validation.
3.13. Disulfide Engineering for Structural Stability of Vaccine Constructs
3.14. Molecular Docking and Interaction of the Multi-Epitope Vaccine Construct with the TLR4 Receptor
3.15. Molecular Dynamic Simulation
3.16. Prediction of Discontinuous B-Cell Epitopes
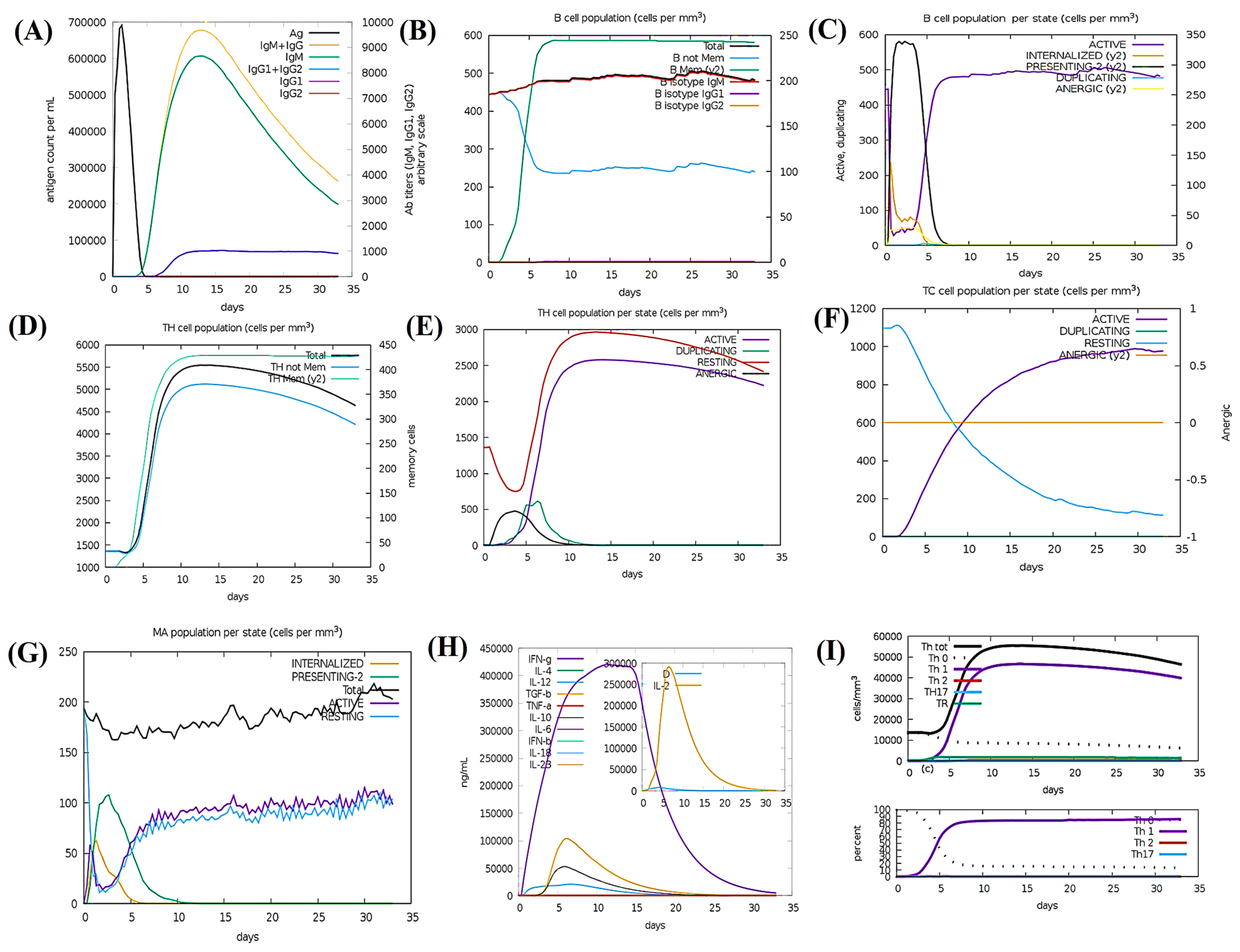
3.17. Simulation of Immunity
3.18. Codon Optimization and Virtual Cloning
3.19. Prediction of mRNA Structure Durability in the Designed Vaccine Construct
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Li, H.; Zhang, X. Cat scratch disease. Radiol. Infect. Dis. 2015, 2, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chen, Y.J.; Tseng, C.S.; Lai, W.L.; Hsu, K.Y.; Chang, C.L.; Lu, C.C.; Hsu, Y.M. A comparative study of the interaction of Bartonella henselae strains with human endothelial cells. Vet. Microbiol. 2011, 149, 147–156. [Google Scholar] [CrossRef]
- Zeaiter, Z.; Fournier, P.E.; Raoult, D. Genomic variation of Bartonella henselae strains detected in lymph nodes of patients with cat scratch disease. J. Clin. Microbiol. 2002, 40, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, N.; Ericson, M.; Maggi, R.; Breitschwerdt, E.B. Vasculitis, cerebral infarction and persistent Bartonella henselae infection in a child. Parasites Vectors 2016, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Mosbacher, M.E.; Klotz, S.; Klotz, J.; Pinnas, J.L. Bartonella henselae and the potential for arthropod vector-borne transmission. Vector-Borne Zoonotic Dis. 2011, 11, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Im, J.-H.; Baek, J.H.; Lee, H.-J.; Lee, J.-S.; Chung, M.-H.; Kim, M.; Lee, S.M.; Kang, J.-S. First case of Bartonella henselae bacteremia in Korea. Infect. Chemother. 2013, 45, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Breitschwerdt, E.B.; Greenberg, R.; Maggi, R.G.; Mozayeni, B.R.; Lewis, A.; Bradley, J.M. Bartonella henselae Bloodstream Infection in a Boy With Pediatric Acute-Onset Neuropsychiatric Syndrome. J. Cent. Nerv. Syst. Dis. 2019, 11, 1179573519832014. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ahmad, S.; Wadood, A.; Rehman, A.U.; Zahid, H.; Qayash Khan, M.; Nawab, J.; Rahman, Z.U.; Alouffi, A.S. Modeling novel putative drugs and vaccine candidates against tick-borne pathogens: A subtractive proteomics approach. Vet. Sci. 2020, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.; Akhtar, S.; Siddiqui, M.H.; Khan, M.K.A. Subtractive proteomics for identification of drug targets in bacterial pathogens: A review. Int. J. Eng. Res. Technol. 2020, 9, 262–273. [Google Scholar]
- Ali, A.; Ahmad, S.; de Albuquerque, P.M.M.; Kamil, A.; Alshammari, F.A.; Alouffi, A.; da Silva Vaz, I., Jr. Prediction of novel drug targets and vaccine candidates against human lice (Insecta), Acari (Arachnida), and their associated pathogens. Vaccines 2021, 10, 8. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Saeed, S.; Munir, S.; Almatroudi, A.; Khurshid, M. In silico subtractive proteomics approach for identification of potential drug targets in Staphylococcus saprophyticus. Int. J. Environ. Res. Public Health 2020, 17, 3644. [Google Scholar] [CrossRef]
- Raju, S.; Rao, V.U.M. Current development strategies for vaccines and the role of reverse vaccinology. Asian J. Pharm. Res. Health Care 2010, 339–346. [Google Scholar]
- de Miguel, N.; Lustig, G.; Twu, O.; Chattopadhyay, A.; Wohlschlegel, J.A.; Johnson, P.J. Proteome analysis of the surface of Trichomonas vaginalis reveals novel proteins and strain-dependent differential expression. Mol. Cell. Proteom. 2010, 9, 1554–1566. [Google Scholar] [CrossRef] [PubMed]
- Esmailnia, E.; Amani, J.; Gargari, S.L.M. Identification of novel vaccine candidate against Salmonella enterica serovar Typhi by reverse vaccinology method and evaluation of its immunization. Genomics 2020, 112, 3374–3381. [Google Scholar] [CrossRef]
- Chakrabarty, R.P.; Alam, A.S.R.; Shill, D.K.; Rahman, A. Identification and qualitative characterization of new therapeutic targets in Stenotrophomonas maltophilia through in silico proteome exploration. Microb. Pathog. 2020, 149, 104293. [Google Scholar] [CrossRef]
- Long, Y.; Sun, J.; Song, T.Z.; Liu, T.; Tang, F.; Zhang, X.; Ding, L.; Miao, Y.; Zhu, W.; Pan, X.; et al. CoVac501, a self-adjuvanting peptide vaccine conjugated with TLR7 agonists, against SARS-CoV-2 induces protective immunity. Cell Discov. 2022, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Mahram, A.; Herbordt, M.C. NCBI BLASTP on high-performance reconfigurable computing systems. ACM Trans. Reconfigurable Technol. Syst. 2015, 7, 1–20. [Google Scholar] [CrossRef]
- Luo, H.; Lin, Y.; Liu, T.; Lai, F.L.; Zhang, C.T.; Gao, F.; Zhang, R. DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res. 2021, 49, D677–D686. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 4, D457–D462. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35 (Suppl. S2), W182–W185. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Genet. 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. 2017 FDA drug approvals. Nat. Rev. Drug Discov. 2018, 17, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Lewis, J.D. Analysis of the human gut microbiome and association with disease. Clin. Gastroenterol. Hepatol. 2013, 11, 774–777. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9. 1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Nielsen, M.; Sette, A. T cell epitope predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Sauna, Z.E. Immunogenicity assessment during the development of protein therapeutics. J. Pharm. Pharmacol. 2018, 70, 584–594. [Google Scholar] [CrossRef]
- Andreatta, M.; Trolle, T.; Yan, Z.; Greenbaum, J.A.; Peters, B.; Nielsen, M. An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics 2018, 34, 1522–1528. [Google Scholar] [CrossRef]
- Nielsen, M.; Lund, O.; Buus, S.; Lundegaard, C. MHC class II epitope predictive algorithms. Immunology 2010, 130, 319–328. [Google Scholar] [CrossRef]
- Těšický, M.; Vinkler, M. Trans-species polymorphism in immune genes: General pattern or MHC-restricted phenomenon? J. Immunol. Res. 2015, 2015, 838035. [Google Scholar] [CrossRef] [PubMed]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 2017, 171, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An introduction to B-cell epitope mapping and in silico epitope prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef] [PubMed]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Empirical predictions of protein conformation. Annu. Rev. Biochem. 1978, 47, 251–276. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef]
- Santhoshkumar, R.; Yusuf, A. In silico structural modeling and analysis of physicochemical properties of curcumin synthase (CURS1, CURS2, and CURS3) proteins of Curcuma longa. J. Genet. Eng. Biotechnol. 2020, 18, 24. [Google Scholar] [CrossRef]
- Zaharieva, N.; Dimitrov, I.; Flower, D.R.; Doytchinova, I. VaxiJen dataset of bacterial immunogens: An update. Curr. Comput. Aided Drug Des. 2019, 15, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, M.; Kolahi, M.; Foroghmand, A.M.; Tabandeh, M.R. In silico assessment of plant L-asparaginase and estimating its allergenicity in comparison to bacteria asparaginase. Issues Hematol. /Oncol. Immunopathol. Pediatr. 2020, 19, 35–46. [Google Scholar] [CrossRef]
- Almofti, Y.A.; Abd-Elrahman, K.A.; Eltilib, E.E. Vaccinomic approach for novel multi epitopes vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). BMC Immunol. 2021, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef]
- Bulaj, G. Formation of disulfide bonds in proteins and peptides. Biotechnol. Adv. 2005, 23, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, A.M.; Caluaco, B.J.; Madureira, J.M.C.; Cabongo, S.Q.; Gaieta, E.M.; Djata, F.; Colares, R.P.; Neto, M.M.; Fernandes, C.F.C.; Marinho, G.S.; et al. Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking, and approach with molecular dynamics, RMSD, RMSF, H-bond, SASA and MMGBSA. Mol. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Min, X.; Li, L.; Yu, H.; Ge, S.; Zhang, J.; Xia, N. Using a machine-learning approach to predict discontinuous antibody-specific B-cell epitopes. Curr. Bioinform. 2017, 12, 406–415. [Google Scholar] [CrossRef]
- Del Tordello, E.; Rappuoli, R.; Delany, I. Reverse vaccinology: Exploiting genomes for vaccine design. In Human Vaccines; Academic Press: Cambridge, MA, USA, 2017; pp. 65–86. [Google Scholar]
- Maity, H.; Wei, A.; Chen, E.; Haidar, J.N.; Srivastava, A.; Goldstein, J. Comparison of predicted extinction coefficients of monoclonal antibodies with experimental values as measured by the Edelhoch method. Int. J. Biol. Macromol. 2015, 77, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.J.; Igwo-Ezikpe, M.; Toye, E.T.; Okpuzor, J. The expression level of a recombinant lipase predicted in silico by different codon optimization algorithms. World Sci. News 2019, 137, 31–41. [Google Scholar]
- Novianti, T. Mutation Detection of Multidrug-Resistant Tuberculosis by RT-PCR Method as the Diagnostic Tool of MDR-TB. J. Bioteknol. Biosains Indones. 2023, 10, 117–127. [Google Scholar]
- Pourseif, M.M.; Parvizpour, S.; Jafari, B.; Dehghani, J.; Naghili, B.; Omidi, Y. A domain-based vaccine construct against SARS-CoV-2, the causative agent of COVID-19 pandemic: Development of self-amplifying mRNA and peptide vaccines. BioImpacts BI 2021, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Rangacharya, O.; Parab, A.; Adkine, S.; Nagargoje, R. A study on the design of an in silico self-amplifying mRNA vaccine against Nipah virus using immunoinformatics. J. Biomol. Struct. Dyn. 2023, 41, 12777–12788. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Kumar, A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. Silico Biol. 2009, 9, 225–231. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T.K. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Zhang, C.; Xia, Y. Identification of genes differentially expressed in vivo by Metarhizium anisopliae in the hemolymph of Locusta migratoria using suppression-subtractive hybridization. Curr. Genet. 2009, 55, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Prosser, G.A.; Larrouy-Maumus, G.; de Carvalho, L.P.S. Metabolomic strategies for the identification of new enzyme functions and metabolic pathways. EMBO Rep. 2014, 15, 657–669. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Contreras, M. Vaccinomics: A future avenue for vaccine development against emerging pathogens. Expert Rev. Vaccines 2021, 20, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Alturki, N.A.; Mashraqi, M.M.; Jalal, K.; Khan, K.; Basharat, Z.; Alzamami, A. Therapeutic target identification and inhibitor screening against riboflavin synthase of colorectal cancer associated fusobacterium nucleatum. Cancers 2022, 14, 6260. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of lipid–protein interactions and lipid-mediated modulation of membrane protein function through molecular simulation. Chem. Rev. 2019, 119, 6086–6161. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kenney, L.J. A new role of OmpR in acid and osmotic stress in Salmonella and E. coli. Front. Microbiol. 2018, 9, 414167. [Google Scholar] [CrossRef]
- Lam, H.; Matroule, J.Y.; Jacobs-Wagner, C. The asymmetric spatial distribution of bacterial signal transduction proteins coordinates cell cycle events. Dev. Cell 2003, 5, 149–159. [Google Scholar] [CrossRef]
- Bedrunka, P. The Role of the Second Messenger Cyclic di-GMP in Bacillus subtilis; Philipps-Universität Marburg: Marburg, Germany, 2017. [Google Scholar]
- Olotu, F.A.; Soliman, M.E. Immunoinformatics prediction of potential B-cell and T-cell epitopes as effective vaccine candidates for eliciting immunogenic responses against Epstein–Barr virus. Biomed. J. 2021, 44, 317–337. [Google Scholar] [CrossRef]
- Sarma, V.R.; Olotu, F.A.; Soliman, M.E. Integrative immunoinformatics paradigm for predicting potential B-cell and T-cell epitopes as viable candidates for subunit vaccine design against COVID-19 virulence. Biomed. J. 2021, 44, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; Reche, P.; Flower, D.R. Selection-based design of in silico dengue epitope ensemble vaccines. Chem. Biol. Drug Des. 2019, 93, 21–28. [Google Scholar] [CrossRef]
- Bettencourt, P.; Müller, J.; Nicastri, A.; Cantillon, D.; Madhavan, M.; Charles, P.D.; Fotso, C.B.; Wittenberg, R.; Bull, N.; Pinpathomrat, N.; et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Zvi, A.; Rotem, S.; Zauberman, A.; Elia, U.; Aftalion, M.; Bar-Haim, E.; Mamroud, E.; Cohen, O. Novel CTL epitopes identified through a Y. pestis proteome-wide analysis in the search for vaccine candidates against plague. Vaccine 2017, 35, 5995–6006. [Google Scholar] [CrossRef] [PubMed]
- Adianingsih, O.R.; Kharisma, V.D. Study of B cell epitope conserved region of the Zika virus envelope glycoprotein to develop multi-strain vaccine. J. Appl. Pharm. Sci. 2019, 9, 98–103. [Google Scholar]
- Pasala, C.; Chilamakuri, C.S.R.; Katari, S.K.; Nalamolu, R.M.; Bitla, A.R.; Amineni, U. Epitope-driven common subunit vaccine design against H. pylori strains. J. Biomol. Struct. Dyn. 2019, 37, 3740–3750. [Google Scholar] [CrossRef] [PubMed]
- Orosco, F.L.; Espiritu, L.M. Navigating the landscape of adjuvants for subunit vaccines: Recent advances and future perspectives. Int. J. Appl. Pharmaceut. 2024, 10, 18–32. [Google Scholar] [CrossRef]
- Chand, Y.; Singh, S. Prioritization of potential vaccine candidates and designing a multiepitope-based subunit vaccine against multidrug-resistant Salmonella Typhi str. CT18: A subtractive proteomics and immunoinformatics approach. Microb. Pathog. 2021, 159, 105150. [Google Scholar] [CrossRef] [PubMed]
- Piri-Gharaghie, T.; Doosti, A.; Mirzaei, S.A. Identification of antigenic properties of Acinetobacter baumannii proteins as novel putative vaccine candidates using reverse vaccinology approach. Appl. Biochem. Biotechnol. 2022, 194, 4892–4914. [Google Scholar] [CrossRef]
- Rawal, K.; Sinha, R.; Nath, S.K.; Preeti, P.; Kumari, P.; Gupta, S.; Sharma, T.; Strych, U.; Hotez, P.; Bottazzi, M.E. Vaxi-DL: A web-based deep learning server to identify potential vaccine candidates. Comput. Biol. Med. 2022, 145, 105401. [Google Scholar] [CrossRef]
- Xiong, W.; Zhang, Q.; Wang, J.; Hao, M.; Zeng, B.; Che, H. Allergenicity evaluation of five types of commercial food-derived oligopeptide products. Food Funct. 2023, 14, 3871–3879. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, M.; Torkzadeh-Mahani, M.; Kargar, F.; Nezafat, N.; Ghasemi, Y. In silico analysis of codon usage and rare codon clusters in the halophilic bacteria L-asparaginase. Biologia 2020, 75, 151–160. [Google Scholar] [CrossRef]
- Suleman, M.; Ul Qamar, M.T.; Kiran; Rasool, S.; Rasool, A.; Albutti, A.; Alsowayeh, N.; Alwashmi, A.S.; Aljasir, M.A.; Ahmad, S.; et al. Immunoinformatics and immunogenetics-based design of immunogenic peptides vaccine against the emerging tick-borne encephalitis virus (Tbev) and its validation through in silico cloning and immune simulation. Vaccines 2021, 9, 1210. [Google Scholar] [CrossRef] [PubMed]
- Kashani-Amin, E.; Tabatabaei-Malazy, O.; Sakhteman, A.; Larijani, B.; Ebrahim-Habibi, A. A systematic review on popularity, application and characteristics of protein secondary structure prediction tools. Curr. Drug Discov. Technol. 2019, 16, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Dhingra, P.; Mishra, A.; Kaushik, R.; Mukherjee, G.; Singh, A.; Shekhar, S. Bhageerath-H: A homology/ab initio hybrid server for predicting tertiary structures of monomeric soluble proteins. BMC Bioinform. 2014, 15, S7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Xue, Y.; Wang, J.; Jia, Z.; Wang, L.; Gong, W. Evaluation of the consistence between the results of immunoinformatics predictions and real-world animal experiments of a new tuberculosis vaccine MP3RT. Front. Cell. Infect. Microbiol. 2022, 12, 1047306. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, H.; Ari, M.M.; Shahlaei, M.; Moradi, S.; Farhadikia, P.; Alvandi, A.; Abiri, R. Designing multi-epitope vaccine against important colorectal cancer (CRC) associated pathogens based on immunoinformatics approach. BMC Bioinform. 2023, 24, 65. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Regar, H.; Verma, V.K.; Prusty, D.; Mishra, A.; Prajapati, V.K. Receptor-ligand based molecular interaction to discover adjuvant for immune cell TLRs to develop next-generation vaccine. Int. J. Biol. Macromol. 2020, 152, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mahmud, S.; Mita, M.A.; Afrose, S.; Hasan, M.R.; Sultana Shimu, M.S.; Saleh, M.A.; Mostafa-Hedeab, G.; Alqarni, M.; Obaidullah, A.J.; et al. Molecular docking and dynamics studies to explore effective inhibitory peptides against the spike receptor binding domain of SARS-CoV-2. Front. Mol. Biosci. 2022, 8, 791642. [Google Scholar] [CrossRef]
- Bahadori, Z.; Shafaghi, M.; Madanchi, H.; Ranjbar, M.M.; Shabani, A.A.; Mousavi, S.F. In silico designing of a novel epitope-based candidate vaccine against Streptococcus pneumoniae with introduction of a new domain of PepO as adjuvant. J. Transl. Med. 2022, 20, 389. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Ferdous, S.; Kelm, S.; Baker, T.S.; Shi, J.; Martin, A.C. B-cell epitopes: Discontinuity and conformational analysis. Mol. Immunol. 2019, 114, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Saleki, K.; Alijanizade, P.; Moradi, S.; Rahmani, A.; Banazadeh, M.; Mohamadi, M.H.; Shahabi, F.; Nouri, H.R. Engineering a novel immunogenic chimera protein utilizing bacterial infections associated with atherosclerosis to induce a deviation in adaptive immune responses via Immunoinformatics approaches. Infect. Genet. Evol. 2022, 102, 105290. [Google Scholar] [CrossRef] [PubMed]
- Fidler, S.; Fox, J.; Tipoe, T.; Longet, S.; Tipton, T.; Abeywickrema, M.; Adele, S.; Alagaratnam, J.; Ali, M.; Aley, P.K.; et al. Booster vaccination against SARS-CoV-2 induces potent immune responses in people with human immunodeficiency virus. Clin. Infect. Dis. 2023, 76, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Sahoo, P.; Mahapatra, S.R.; Dey, J.; Ghosh, M.; Kushwaha, G.S.; Misra, N.; Suar, M.; Raina, V.; Son, Y.O. Development of a conserved chimeric vaccine for induction of strong immune response against Staphylococcus aureus using immunoinformatics approaches. Vaccines 2021, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Shams, M.; Heydaryan, S.; Bashi, M.C.; Gorgani, B.N.; Ghasemi, E.; Majidiani, H.; Nazari, N.; Irannejad, H. In silico design of a novel peptide-based vaccine against the ubiquitous apicomplexan Toxoplasma gondii using surface antigens. Silico Pharmacol. 2023, 11, 5. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Awais, M.; Aldhahrani, A.; Althobaiti, F.; Alhazmi, A.; Sattar, S.; Afzal, U.; Baeshen, H.A.; El Enshasy, H.A.; Dailin, D.J.; et al. Designing a highly immunogenic multi epitope based subunit vaccine against Bacillus cereus. Saudi J. Biol. Sci. 2021, 28, 4859–4866. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Patra, P.; Ghosh, P.; Sharma, G.; Patra, B.C.; Saha, R.P.; Lee, S.S.; Chakraborty, C. A SARS-CoV-2 vaccine candidate: In-silico cloning and validation. Inform. Med. Unlocked 2020, 20, 100394. [Google Scholar] [CrossRef] [PubMed]
- Rouse, C.J.; Hawkins, K.; Kabbej, N.; Dalugdug, J.; Kunta, A.; Kim, M.J.; Someya, S.; Herbst, Z.; Gelb, M.; Dinelli, I.; et al. Disease correction in mucopolysaccharidosis type IIIB mice by intraparenchymal or cisternal delivery of a capsid modified AAV8 codon-optimized NAGLU vector. Hum. Mol. Genet. 2023, 32, 417–430. [Google Scholar] [CrossRef]
- Sato, K.; Hamada, M.; Asai, K.; Mituyama, T. CENTROIDFOLD: A web server for RNA secondary structure prediction. Nucleic Acids Res. 2009, 37 (Suppl. S2), W277–W280. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Subtractive Approaches | B. henselae Strain Houston-1 |
|---|---|---|
| 1 | Complete set of proteins | 1481 |
| 2 | Mini proteins | 268 |
| 3 | Paralogous proteins in CD-HIT | 1213 |
| 4 | Non-homologs | 827 |
| 5 | Vital proteins in DEG | 153 |
| 6 | Unique metabolic pathways at KEGG | 24 |
| 7 | Number of vital proteins involved in KEGG and KAAS | 20 |
| 8 | Druggable proteins | 9 |
| 9 | Gut flora proteins | 6 |
| 10 | Cytoplasmic proteins | 5 |
| 11 | Membrane protein | 1 |
| Protein ID | Protein Name | Drugbank ID | Chemical Formula | Drug Name | Drug Group | Drugbank Organism | Localization | Virulency | Antigenicity Score | Antigenicity | Allergenicity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WP_011180971.1 | UDP-N-acetylmuramate--L-alanine ligase | DB01673 DB03909 DB04395 | C23H36N4O20P2 C11H18N5O12P3 C10H17N6O12P3 | Uridine-5’-Diphosphate-N-Acetylmuramoyl-L-Alanine Adenosine-5’- [beta, Gamma-methylene]triphosphate Phosphoaminophosphonic Acid-Adenylate Ester | Experimental Experimental Experimental | Haemophilus influenzae (strain ATCC 51,907/DSM 11,121/KW20/Rd) | Cytoplasmic | Non-Virulent | 0.3964 | Non-Antigenic | Non-Allergic |
| WP_011180187.1 | 3-deoxy-manno-octulosonate cytidylyltransferase | DB04482 | C17H26N3O15P | Cmp-2-Keto-3-Deoxy-Octulosonic Acid | Experimental | Haemophilus influenzae (strain ATCC 51,907/DSM 11,121/KW20/Rd) | Cytoplasmic | Virulent | 0.3278 | Non-Antigenic | Non-Allergic |
| WP_011180414.1 | PAS domain-containing sensor histidine kinase (ATP-binding protein) | DB02071 DB03366 | C4H7N2 C3H4N2 | 1-Methylimidazole Imidazole | Experimental Experimental Investigational | Bradyrhizobium diazofficiens strain (JCM 10833/AM 13628) | Membrane-Bound | Virulent | 0.4060 | Antigenic | Non-Allergic |
| WP_011180514.1 | sigma-54-dependent Fis family transcriptional regulator | DB01857 | C4H8NO7P | Phosphoaspartate | Experimental | Salmonella Typhimurium strain (CT2 1412/ATCC 700720) | Cytoplasmic | Virulent | 0.3676 | Non-Antigenic | Non-Allergic |
| WP_011180500.1 | 3-deoxy-8-phosphooctulonate synthase | DB01819 DB02433 DB03113 DB03936 | C3H5O6P C9H23NO13P2 C3H6FO6P C5H11O7P | Phosphoenolpyruvate {[(2,2-Dihydroxy-Ethyl) -(2,3,4,5-Tetrahydroxy-6-Phosphonooxy-Hexyl)-Amino]-Methyl}-Phosphonic Acid 3-Fluoro-2-(Phosphonooxy)Propanoic Acid 1-Deoxy-Ribofuranose-5’-Phosphate | Experimental Experimental Experimental Experimental | Shigella flexneri | Cytoplasmic | Virulent | 0.3274 | Non-Antigenic | Non-Allergic |
| WP_034454605.1 | Phosphoenolpyruvate--protein phosphotransferase | DB08357 | C8H18O3 | Diethylene glycol diethyl ether | Experimental | Acinetobacter baylyi strain ATCC 33305/ADP1) | Cytoplasmic | Non-Virulent | 0.3740 | Non-Antigenic | Non-Allergic |
| T-Cell Epitopes | Antigenicity Score | Allergenicity | Toxicity | SVM | Class I Immunogenicity |
|---|---|---|---|---|---|
| AAIRFVSIY | 0.8849 (Antigenic) | Non-Allergic | Non-toxic | −1.36 | 0.18628 |
| ILALLYAYY | 1.2102 (Antigenic) | Non-Allergic | Non-toxic | −0.77 | 0.01812 |
| VTDEEELHL | 1.0484 (Antigenic) | Non-Allergic | Non-toxic | −0.65 | 0.30924 |
| TADGCWLKI | 0.7581 (Antigenic) | Non-Allergic | Non-toxic | −0.21 | 0.06195 |
| MHC-II Peptide | Start | HLA Alleles | Antigenicity Score | Allergenicity | Toxicity |
|---|---|---|---|---|---|
| ALLYAYYKTDSISEK | 39 | HLA-DRB1 * 04:05 | 0.4707 (Antigenic) | Non-Allergic | Non-Toxic |
| IALSHTYISEKTQEI | 21 | HLA-DRB3 * 01:01 | 0.4331 (Antigenic) | Non-Allergic | Non-Toxic |
| KTDSISEKIRAMYEM | 46 | HLA-DRB1 * 13:02 | 0.6838 (Antigenic) | Non-Allergic | Non-Toxic |
| LLYAYYKTDSISEKI | 40 | HLA-DRB1 * 04:05 | 0.5207 (Antigenic) | Non-Allergic | Non-Toxic |
| YKTDSISEKIRAMYE | 45 | HLA-DRB1 * 13:02 | 0.4995 (Antigenic) | Non-Allergic | Non-Toxic |
| B-Cell Peptide | Antigenicity Score | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| KSSAQNHKARTKHINP | 0.6231 | Antigenic | Non-Allergic | Non-toxic |
| TSIRQTADGCWLKINE | 0.6391 | Antigenic | Non-Allergic | Non-toxic |
| LAAIRFVSIYDLRHTI | 0.6559 | Antigenic | Non-Allergic | Non-toxic |
| KLEIISKEMKGTTVTI | 0.6320 | Antigenic | Non-Allergic | Non-toxic |
| KEMKGTTVTITMPIKQ | 0.4484 | Antigenic | Non-Allergic | Non-toxic |
| NQLTKTHTGSGLGLAI | 1.0892 | Antigenic | Non-Allergic | Non-toxic |
| RHTIDKNTRSTITLLA | 2.2375 | Antigenic | Non-Allergic | Non-toxic |
| Physiochemical Features | Evaluation |
|---|---|
| Amino acid residue | 367 |
| Molecular weight | 38.84 kDa |
| Theoretical PI | 9.34 |
| Total number of negatively charged residue (Asp + Glu) | 28 |
| Total number of positively charged residues (Arg + Lys) | 44 |
| Formula | C1737H2748N472O509S13 |
| Extinction coefficients | 97,000 M−1 Cm−1 |
| Estimated half-life | 30 h (mammalian reticulocytes, in vitro) >20 h (Yeast, in vitro) >10 h (Escherichiaa coli, in vivo) |
| Instability index | 32.68 (stable) |
| Aliphatic index | 75.59 |
| Gravy | −0.377 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Chiou, C.-C.; Ahmad, S.; Islam, Z.U.; Tanaka, T.; Alouffi, A.; Chen, C.-C.; Almutairi, M.M.; Ali, A. Subtractive Proteomics and Reverse-Vaccinology Approaches for Novel Drug Target Identification and Chimeric Vaccine Development against Bartonella henselae Strain Houston-1. Bioengineering 2024, 11, 505. https://doi.org/10.3390/bioengineering11050505
Rahman S, Chiou C-C, Ahmad S, Islam ZU, Tanaka T, Alouffi A, Chen C-C, Almutairi MM, Ali A. Subtractive Proteomics and Reverse-Vaccinology Approaches for Novel Drug Target Identification and Chimeric Vaccine Development against Bartonella henselae Strain Houston-1. Bioengineering. 2024; 11(5):505. https://doi.org/10.3390/bioengineering11050505
Chicago/Turabian StyleRahman, Sudais, Chien-Chun Chiou, Shabir Ahmad, Zia Ul Islam, Tetsuya Tanaka, Abdulaziz Alouffi, Chien-Chin Chen, Mashal M. Almutairi, and Abid Ali. 2024. "Subtractive Proteomics and Reverse-Vaccinology Approaches for Novel Drug Target Identification and Chimeric Vaccine Development against Bartonella henselae Strain Houston-1" Bioengineering 11, no. 5: 505. https://doi.org/10.3390/bioengineering11050505