
Genome-Wide Identification and Functional Analysis of Salvia miltiorrhiza MicroRNAs Reveal the Negative Regulatory Role of Smi-miR159a in Phenolic Acid Biosynthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. High-Throughput Sequencing of S. miltiorrhiza sRNAs
2.2. Identification of Known and Novel miRNAs in S. miltiorrhiza
2.3. Expression Patterns of S. miltiorrhiza miRNAs
2.4. Identification of miRNA Targets in S. miltiorrhiza
2.5. Expression Analysis and 5′ RLM-RACE Validation of Smi-miR159 Targets
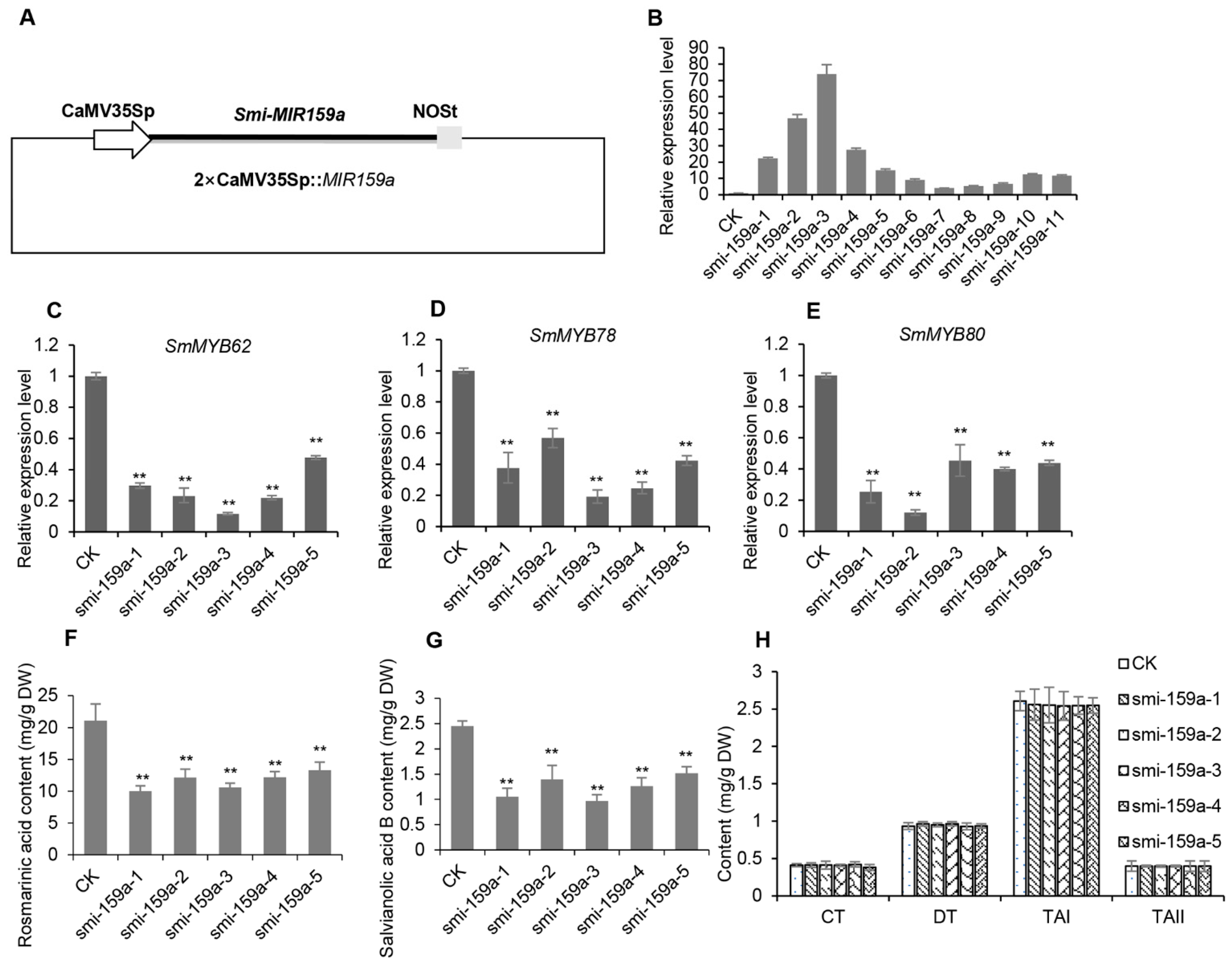
2.6. Smi-miR159a Regulates the Biosynthesis of RA and SalB but Not Tanshinones in S. miltiorrhiza
3. Materials and Methods
3.1. Plant Materials and Sample Collection
3.2. sRNA and Degradome Sequencing
3.3. Data Processing and Computational Identification of miRNAs
3.4. Prediction of miRNAs Targets and Degradome Analysis
3.5. Experimental Verification of miRNA-Directed Target Cleavage
3.6. Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
3.7. Plasmid Construction and Transformation
3.8. UPLC Determination of RA and SalB
3.9. UPLC Determination of Tanshinones
3.10. Expression Analysis of Key Enzyme Genes
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.; Martienssen, R.A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 2015, 16, 727–741. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, B.; Zhao, D.; Li, C.; Shao, F.; Lu, S. Genome-wide analysis and molecular dissection of the SPL gene family in Salvia miltiorrhiza. J. Integr. Plant Biol. 2014, 56, 38–50. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, X.; Shi, M.; Yu, J.; Guo, C. The miR159-MYB33-ABI5 module regulates seed germination in Arabidopsis. Physiol. Plant 2022, 174, e13659. [Google Scholar] [CrossRef]
- Achard, P.; Herr, A.; Baulcombe, D.C.; Harberd, N.P. Modulation of floral development by a gibberellin-regulated microRNA. Development 2004, 131, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Vallarino, J.G.; Osorio, S.; Bombarely, A.; Casañal, A.; Cruz-Rus, E.; Sánchez-Sevilla, J.F.; Amaya, I.; Giavalisco, P.; Fernie, A.R.; Botella, M.A.; et al. Central role of FaGAMYB in the transition of the strawberry receptacle from development to ripening. New Phytol. 2015, 208, 482–496. [Google Scholar]
- Wang, C.; Jogaiah, S.; Zhang, W.; Abdelrahman, M.; Fang, J.G. Spatio-temporal expression of miRNA159 family members and their GAMYB target gene during the modulation of gibberellin-induced grapevine parthenocarpy. J. Exp. Bot. 2018, 69, 3639–3650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wang, F.; Deng, Y.; Zhong, F.; Tian, P.; Lin, D.; Deng, J.; Zhang, Y.; Huang, T. Sly-miR159 regulates fruit morphology by modulating GA biosynthesis in tomato. Plant Biotechnol. J. 2022, 20, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Bussell, J.D.; Pacurar, D.I.; Schwambach, J.; Pacurar, M.; Bellini, C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 2009, 21, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of MIRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Tirumalai, V.; Swetha, C.; Nair, A.; Pandit, A.; Shivaprasad, P.V. miR828 and miR858 regulate VvMYB114 to promote anthocyanin and flavonol accumulation in grapes. J. Exp. Bot. 2019, 70, 4775–4792. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Badola, P.K.; Bhatia, C.; Sharma, D.; Trivedi, P.K. Primary transcript of miR858 encodes regulatory peptide and controls flavonoid biosynthesis and development in Arabidopsis. Nat. Plants 2020, 6, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, Q.; Wei, H.; Chang, M.J.; Tunlaya-Anukit, S.; Kim, H.; Liu, J.; Song, J.; Sun, Y.H.; Yuan, L.; et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2013, 110, 10848–11853. [Google Scholar] [CrossRef]
- Cheng, T.O. Danshen: A popular Chinese cardiac herbal drug. J. Am. Coll. Cardiol. 2006, 47, 1498. [Google Scholar] [CrossRef]
- Cheng, T.O. Cardiovascular effects of Danshen. Int. J. Cardiol. 2007, 121, 9–22. [Google Scholar] [CrossRef]
- He, X.; Chen, Y.; Xia, Y.; Hong, X.; You, H.; Zhang, R.; Liang, Z.; Cui, Q.; Zhang, S.; Zhou, M.; et al. DNA methylation regulates biosynthesis of tanshinones and phenolic acids during growth of Salvia miltiorrhiza. Plant Physiol. 2024, 194, 2086–2100. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.; Deng, Y.; Wei, H.; Lu, S. Characteristics of Salvia miltiorrhiza methylome and the regulatory mechanism of DNA methylation in tanshinone biosynthesis. Hortic. Res. 2023, 10, uhad114. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Xie, Y.; Zhang, Y.; Cai, X.; Zhang, B.; Ma, P.; Dong, J. Salicylic acid regulates phenolic acid biosynthesis via SmNPR1-SmTGA2/SmNPR4 modules in Salvia miltiorrhiza. J. Exp. Bot. 2023, 74, 5736–5751. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Zhang, B.; Zhang, S.; Hao, R.; Xia, Y.; Ma, P.; Dong, J. The SmNPR4-SmTGA5 module regulates SA-mediated phenolic acid biosynthesis in Salvia miltiorrhiza hairy roots. Hortic. Res. 2023, 10, uhad066. [Google Scholar] [CrossRef]
- Liu, S.; Gao, X.; Shi, M.; Sun, M.; Li, K.; Cai, Y.; Chen, C.; Wang, C.; Maoz, I.; Guo, X.; et al. Jasmonic acid regulates the biosynthesis of medicinal metabolites via the JAZ9-MYB76 complex in Salvia miltiorrhiza. Hortic. Res. 2023, 10, uhad004. [Google Scholar] [CrossRef]
- Li, Q.; Fang, X.; Zhao, Y.; Cao, R.; Dong, J.; Ma, P. The SmMYB36-SmERF6/SmERF115 module regulates the biosynthesis of tanshinones and phenolic acids in Salvia miltiorrhiza hairy roots. Hortic. Res. 2022, 10, uhac238. [Google Scholar] [CrossRef]
- Ma, Y.; Cui, G.; Chen, T.; Ma, X.; Wang, R.; Jin, B.; Yang, J.; Kang, L.; Tang, J.; Lai, C.; et al. Expansion within the CYP71D subfamily drives the heterocyclization of tanshinones synthesis in Salvia miltiorrhiza. Nat. Commun. 2021, 12, 685. [Google Scholar] [CrossRef]
- Guo, J.; Ma, X.; Cai, Y.; Ma, Y.; Zhan, Z.; Zhou, Y.J.; Liu, W.; Guan, M.; Yang, J.; Cui, G.; et al. Cytochrome P450 promiscuity leads to a bifurcating biosynthetic pathway for tanshinones. New Phytol. 2016, 210, 525–534. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, Y.J.; Hillwig, M.L.; Shen, Y.; Yang, L.; Wang, Y.; Zhang, X.; Liu, W.; Peters, R.J.; Chen, X.; et al. CYP76AH1 catalyzes turnover of miltiradiene in tanshinones biosynthesis and enables heterologous production of ferruginol in yeasts. Proc. Natl. Acad. Sci. USA 2013, 110, 12108–12113. [Google Scholar] [CrossRef]
- Li, C.; Li, D.; Li, J.; Shao, F.; Lu, S. Characterization of the polyphenol oxidase gene family reveals a novel microRNA involved in posttranscriptional regulation of PPOs in Salvia miltiorrhiza. Sci. Rep. 2017, 7, 44622. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, C.; Qiu, X.; Lu, S. Systematic analysis of alkaline/neutral invertase genes reveals the involvement of Smi-miR399 in regulation of SmNINV3 and SmNINV4 in Salvia miltiorrhiza. Plants 2019, 8, 490. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, H.; Chen, M.; Zhang, J.; Tan, R.; Zhao, S.; Wang, Z. Smi-miR396b targeted SmGRFs, SmHDT1, and SmMYB37/4 synergistically regulates cell growth and active ingredient accumulation in Salvia miltiorrhiza hairy roots. Plant Cell Rep. 2020, 39, 1263–1283. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Guo, X.; Yang, R.; Wang, S.; Li, L.; Niu, J.; Wang, D.; Cao, X. MiR408-SmLAC3 module participates in salvianolic acid B synthesis in Salvia miltiorrhiza. Int. J. Mol. Sci. 2021, 22, 7541. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, M.; Pang, Y.; Hu, X.; Sun, C.; Zhou, H.; Deng, Y.; Lu, S. The smi–miR858a-SmMYB module regulates tanshinone and phenolic acid biosynthesis in Salvia miltiorrhiza. Hortic. Res. 2024, 11, uhae047. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, Q.; Ma, X.; Ying, Q.; Shen, B.; Qian, Y.; Song, H.; Wang, H. Deep sequencing identifies tissue-specific microRNAs and their target genes involving in the biosynthesis of tanshinones in Salvia miltiorrhiza. PLoS ONE 2014, 9, e111679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, W.; Zhu, X.; Liu, L.; He, Z.; Yang, S.; Liang, Z.; Yan, X.; He, Y.; Liu, Y. Identification and characterization of Salvia miltiorrhizain miRNAs in response to replanting disease. PLoS ONE 2016, 11, e0159905. [Google Scholar] [CrossRef]
- Zhang, G.; Tian, Y.; Zhang, J.; Shu, L.; Yang, S.; Wang, W.; Sheng, J.; Dong, Y.; Chen, W. Hybrid de novo genome assembly of the Chinese herbal plant danshen (Salvia miltiorrhiza Bunge). Gigascience 2015, 4, 62. [Google Scholar] [CrossRef]
- Xu, H.; Song, J.; Luo, H.; Zhang, Y.; Li, Q.; Zhu, Y.; Xu, J.; Li, Y.; Song, C.; Wang, B.; et al. Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant 2016, 9, 949–952. [Google Scholar] [CrossRef]
- Song, Z.; Lin, C.; Xing, P.; Fen, Y.; Jin, H.; Zhou, C.; Gu, Y.Q.; Wang, J.; Li, X. A high-quality reference genome sequence of Salvia miltiorrhiza provides insights into tanshinone synthesis in its red rhizomes. Plant Genome 2020, 13, e20041. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chang, Y.; Li, C.; Qiu, X.; Cui, X.; Meng, F.; Zhang, S.; Li, X.; Lu, S. Chromosome-level genome assembly of Salvia miltiorrhiza with orange roots uncovers the role of Sm2OGD3 in catalyzing 15,16-dehydrogenation of tanshinones. Hortic. Res. 2023, 10, uhad069. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Park, S.; Zhai, J.; Gurazada, S.G.; De Paoli, E.; Meyers, B.C.; Green, P.J. Massive analysis of rice small RNAs: Mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23, 4185–4207. [Google Scholar] [CrossRef] [PubMed]
- Breakfield, N.W.; Corcoran, D.L.; Petricka, J.J.; Shen, J.; Sae-Seaw, J.; Rubio-Somoza, I.; Weigel, D.; Ohler, U.; Benfey, P.N. High-resolution experimental and computational profiling of tissue-specific known and novel miRNAs in Arabidopsis. Genome Res. 2012, 22, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.G.; Park, J.H.; Lim, J.Y.; Kim, D.; Choi, Y.; Kim, S.; Reeves, G.; Yeom, S.I.; Lee, J.S.; Park, M.; et al. The hot pepper Capsicum annuum microRNA transcriptome reveals novel and conserved targets: A foundation for understanding MicroRNA functional roles in hot pepper. PLoS ONE 2013, 8, e64238. [Google Scholar] [CrossRef] [PubMed]
- Klevebring, D.; Street, N.R.; Fahlgren, N.; Kasschau, K.D.; Carrington, J.C.; Lundeberg, J.; Jansson, S. Genome-wide profiling of populus small RNAs. BMC Genom. 2009, 10, 620. [Google Scholar] [CrossRef]
- Wu, B.; Wang, M.; Ma, Y.; Yuan, L.; Lu, S. High-throughput sequencing and characterization of the small RNA transcriptome reveal features of novel and conserved microRNAs in Panax ginseng. PLoS ONE 2012, 7, e44385. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes. Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Zhu, H.; An, Y.Q.; Beers, E.P.; Liu, Z. Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 2012, 13, R47. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lu, S. Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genom. 2014, 15, 277. [Google Scholar] [CrossRef]
- Shao, F.; Lu, S. Genome-wide identification, molecular cloning, expression profiling and posttranscriptional regulation analysis of the Argonaute gene family in Salvia miltiorrhiza, an emerging model medicinal plant. BMC Genom. 2013, 14, 512. [Google Scholar] [CrossRef]
- Wang, B.; Sun, W.; Li, Q.; Li, Y.; Luo, H.; Song, J.; Sun, C.; Qian, J.; Zhu, Y.; Hayward, A.; et al. Genome-wide identification of phenolic acid biosynthetic genes in Salvia miltiorrhiza. Planta 2015, 241, 711–725. [Google Scholar] [CrossRef]
- Lu, S. Biosynthesis and regulatory mechanisms of bioactive compounds in Salvia miltiorrhiza, a model system for medicinal plant biology. Crit. Rev. Plant Sci. 2021, 40, 243–283. [Google Scholar] [CrossRef]
- Yan, C.; Li, C.; Jiang, M.; Xu, Y.; Zhang, S.; Hu, X.; Chen, Y.; Lu, S. Systematic characterization of gene families and functional analysis of PvRAS3 and PvRAS4 involved in rosmarinic acid biosynthesis in Prunella vulgaris. Front. Plant Sci. 2024, 15, 1374912. [Google Scholar] [CrossRef]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef]
- Ma, Z.; Coruh, C.; Axtell, M.J. Arabidopsis lyrata small RNAs: Transient MIRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell 2010, 22, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Lu, S.; Sun, Y.H.; Shi, R.; Clark, C.; Li, L.; Chiang, V.L. Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 2005, 17, 2186–2203. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Jiang, M.; Li, J.; Xu, Y.; Li, C.; Lu, S. Genome-Wide Identification and Functional Analysis of Salvia miltiorrhiza MicroRNAs Reveal the Negative Regulatory Role of Smi-miR159a in Phenolic Acid Biosynthesis. Int. J. Mol. Sci. 2024, 25, 5148. https://doi.org/10.3390/ijms25105148
Zhou H, Jiang M, Li J, Xu Y, Li C, Lu S. Genome-Wide Identification and Functional Analysis of Salvia miltiorrhiza MicroRNAs Reveal the Negative Regulatory Role of Smi-miR159a in Phenolic Acid Biosynthesis. International Journal of Molecular Sciences. 2024; 25(10):5148. https://doi.org/10.3390/ijms25105148
Chicago/Turabian StyleZhou, Hong, Maochang Jiang, Jiang Li, Yayun Xu, Caili Li, and Shanfa Lu. 2024. "Genome-Wide Identification and Functional Analysis of Salvia miltiorrhiza MicroRNAs Reveal the Negative Regulatory Role of Smi-miR159a in Phenolic Acid Biosynthesis" International Journal of Molecular Sciences 25, no. 10: 5148. https://doi.org/10.3390/ijms25105148