

Modulation of Serotonin-Related Genes by Extracellular Vesicles of the Probiotic Escherichia coli Nissle 1917 in the Interleukin-1β-Induced Inflammation Model of Intestinal Epithelial Cells
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Modulation of the Serotonergic System by EcN or EcoR12 EVs
2.2. Regulation of miRNAs That Target SERT mRNA by EcN or EcoR12 EVs
2.3. Modulation of Innate Immune Receptors by EcN or EcoR12 EVs
2.4. Modulation of Pro-Inflammatory and Oxidative Stress Markers by EcN or EcoR12 EVs
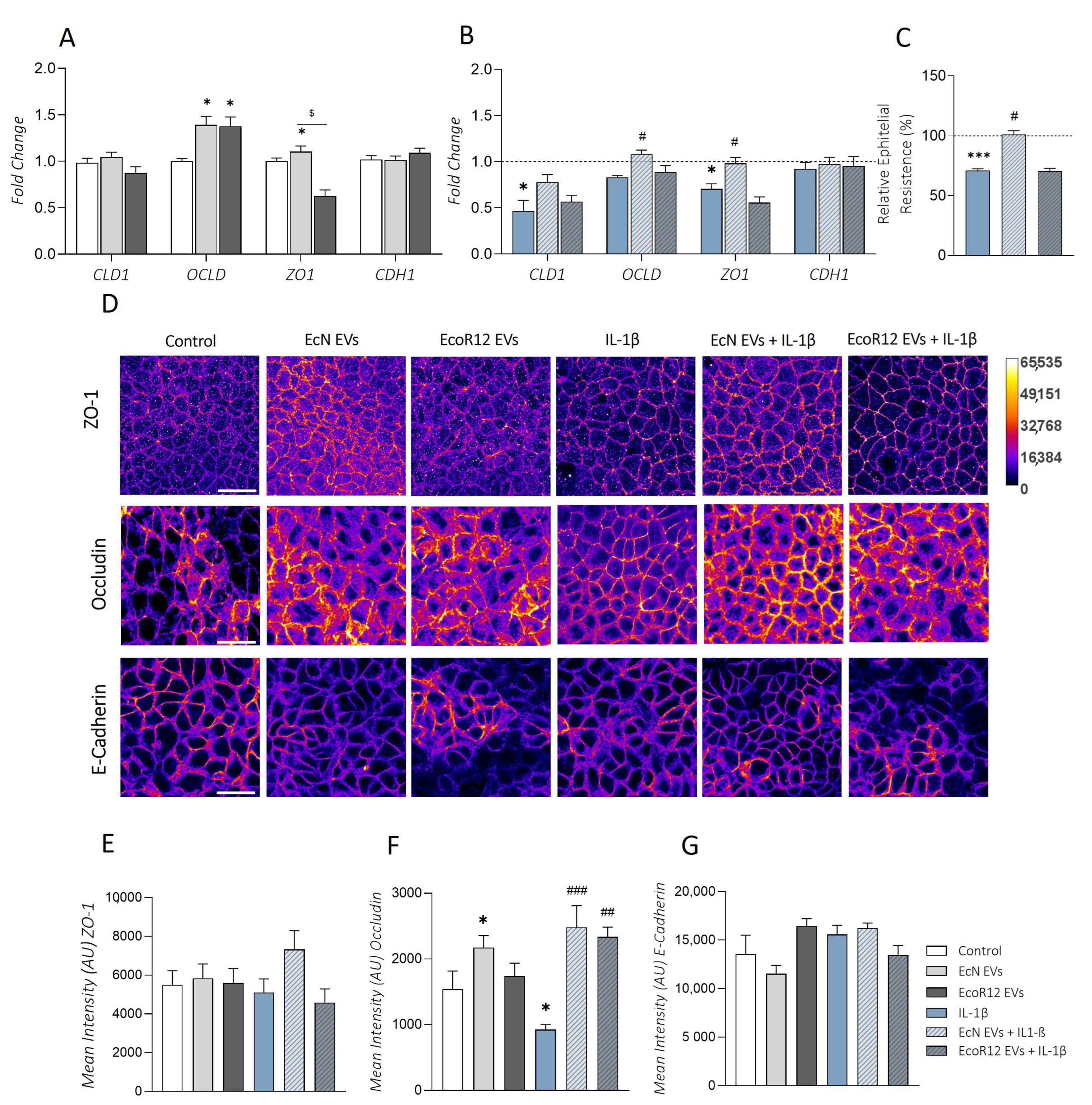
2.5. Effects of EcN or EcoR12 EVs on IL-1β-Induced Epithelial Barrier Damage
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Isolation of EVs
4.2. Cell Culture and Stimulation Conditions
4.3. Cell Viability Assays
4.4. Measurement of Transepithelial Electrical Resistance (TER)
4.5. RNA Extraction and Quantitative Reverse Transcription–Polymerase Chain Reaction (RT-qPCR)
4.6. Quantification of Cytokines and Serotonin (5-HT) through ELISA
4.7. Immunofluorescence Labelling
4.8. Confocal Microscopy
4.9. Image Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the Normal Gut Microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Roles of Intestinal Epithelial Cells in the Maintenance of Gut Homeostasis. Exp. Mol. Med. 2017, 49, e338–e345. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Garrido, N.; Badia, J.; Baldomà, L. Microbiota-derived Extracellular Vesicles in Interkingdom Communication in the Gut. J. Extracell. Vesicles 2021, 10, e12161. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut Microbiome and Liver Diseases. Gut 2016, 65, 2035–2044. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut Microbiome and Health: Mechanistic Insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- Koopman, N.; Katsavelis, D.; Ten Hove, A.; Brul, S.; de Jonge, W.; Seppen, J. The Multifaceted Role of Serotonin in Intestinal Homeostasis. Int. J. Mol. Sci. 2021, 22, 9487. [Google Scholar] [CrossRef] [PubMed]
- Nzakizwanayo, J.; Dedi, C.; Standen, G.; Macfarlane, W.M.; Patel, B.A.; Jones, B.V. Escherichia Coli Nissle 1917 Enhances Bioavailability of Serotonin in Gut Tissues through Modulation of Synthesis and Clearance. Sci. Rep. 2015, 5, 17324. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubfar, R.; Behrouzi, A.; Zare Banadkoki, E.; Ashrafian, F.; Lari, A.; Vaziri, F.; Nojoumi, S.A.; Fateh, A.; Khatami, S.; Siadat, S.D. Effect of Akkermansia Muciniphila, Faecalibacterium Prausnitzii, and Their Extracellular Vesicles on the Serotonin System in Intestinal Epithelial Cells. Probiotics Antimicrob. Proteins 2021, 13, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubfar, R.; Behrouzi, A.; Ashrafian, F.; Shahryari, A.; Moradi, H.R.; Choopani, S.; Hadifar, S.; Vaziri, F.; Nojoumi, S.A.; Fateh, A.; et al. Modulation of Serotonin Signaling/Metabolism by Akkermansia Muciniphila and Its Extracellular Vesicles through the Gut-Brain Axis in Mice. Sci. Rep. 2020, 10, 22119. [Google Scholar] [CrossRef] [PubMed]
- Layunta, E.; Latorre, E.; Forcén, R.; Grasa, L.; Plaza, M.A.; Arias, M.; Alcalde, A.I.; Mesonero, J.E. NOD1 Downregulates Intestinal Serotonin Transporter and Interacts with Other Pattern Recognition Receptors. J. Cell Physiol. 2018, 233, 4183–4193. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Layunta, E.; Grasa, L.; Castro, M.; Pardo, J.; Gomollón, F.; Alcalde, A.I.; Mesonero, J.E. Intestinal Serotonin Transporter Inhibition by Toll-Like Receptor 2 Activation. A Feedback Modulation. PLoS ONE 2016, 11, e0169303. [Google Scholar] [CrossRef] [PubMed]
- Qasem, A.; Naser, A.E.; Naser, S.A. Enteropathogenic Infections Modulate Intestinal Serotonin Transporter (SERT) Function by Activating Toll-like Receptor 2 (TLR-2) in Crohn’s Disease. Sci. Rep. 2021, 11, 22624. [Google Scholar] [CrossRef]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl Hydrocarbon Receptor and Intestinal Immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.D.; Tekin, I.; Vrana, K.E.; Mawe, G.M. Review Article: The Many Potential Roles of Intestinal Serotonin (5-Hydroxytryptamine, 5-HT) Signalling in Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2017, 46, 569–580. [Google Scholar] [CrossRef] [PubMed]
- González Delgado, S.; Garza-Veloz, I.; Trejo-Vazquez, F.; Martinez-Fierro, M.L. Interplay between Serotonin, Immune Response, and Intestinal Dysbiosis in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 5632. [Google Scholar] [CrossRef] [PubMed]
- Jørandli, J.W.; Thorsvik, S.; Skovdahl, H.K.; Kornfeld, B.; Sæterstad, S.; Gustafsson, B.I.; Sandvik, A.K.; Van Beelen Granlund, A. The Serotonin Reuptake Transporter Is Reduced in the Epithelium of Active Crohn’s Disease and Ulcerative Colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, 761–768. [Google Scholar] [CrossRef]
- Datta, N.; Johnson, C.; Kao, D.; Gurnani, P.; Alexander, C.; Polytarchou, C.; Monaghan, T.M. MicroRNA-Based Therapeutics for Inflammatory Disorders of the Microbiota-Gut-Brain Axis. Pharmacol. Res. 2023, 194, 6870. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, A.; Rankin, C.R.; Polytarchou, C.; Lokhandwala, Z.A.; Patel, A.; Chang, L.; Pothoulakis, C.; Iliopoulos, D.; Padua, D.M. MiR-24 Is Elevated in Ulcerative Colitis Patients and Regulates Intestinal Epithelial Barrier Function. Am. J. Pathol. 2019, 189, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Wang, H.; Denou, E.; Ghia, J.E.; Rossi, L.; Fontes, M.E.; Bernier, S.P.; Shajib, M.S.; Banskota, S.; Collins, S.M.; et al. Modulation of Gut Microbiota Composition by Serotonin Signaling Influences Intestinal Immune Response and Susceptibility to Colitis. CMGH 2019, 7, 709–728. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Tong, X.; Wang, R.; Song, X. The Clinical Effects of Probiotics for Inflammatory Bowel Disease. Medicine 2018, 97, e13792. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Esposito, A.; Ercolini, D. Outlook on Next-Generation Probiotics from the Human Gut. Cell. Mol. Life Sci. 2022, 79, 14–18. [Google Scholar] [CrossRef]
- Ma, L.; Tu, H.; Chen, T. Postbiotics in Human Health: A Narrative Review. Nutrients 2023, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Salminen, S.; Collado, M.C.; Endo, A.; Hill, C.; Lebeer, S.; Quigley, E.M.M.; Sanders, M.E.; Shamir, R.; Swann, J.R.; Szajewska, H.; et al. The International Scientific Association of Probiotics and Prebiotics (ISAPP) Consensus Statement on the Definition and Scope of Postbiotics. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 649–667. [Google Scholar] [CrossRef] [PubMed]
- González-Lozano, E.; García-García, J.; Gálvez, J.; Hidalgo-García, L.; Rodríguez-Nogales, A.; Rodríguez-Cabezas, M.E.; Sánchez, M. Novel Horizons in Postbiotics: Lactobacillaceae Extracellular Vesicles and Their Applications in Health and Disease. Nutrients 2022, 14, 5296. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, Q.; Nie, S. Bacterial Extracellular Vesicles: An Emerging Postbiotic. Trends Food Sci. Technol. 2024, 143, 104275. [Google Scholar] [CrossRef]
- Alvarez, C.-S.; Badia, J.; Bosch, M.; Giménez, R.; Baldomà, L. Outer Membrane Vesicles and Soluble Factors Released by Probiotic Escherichia Coli Nissle 1917 and Commensal ECOR63 Enhance Barrier Function by Regulating Expression of Tight Junction Proteins in Intestinal Epithelial Cells. Front. Microbiol. 2016, 7, 1981. [Google Scholar] [CrossRef]
- Olivo-Martínez, Y.; Bosch, M.; Badia, J.; Baldomà, L. Modulation of the Intestinal Barrier Integrity and Repair by Microbiota Extracellular Vesicles through the Differential Regulation of Trefoil Factor 3 in LS174T Goblet Cells. Nutrients 2023, 15, 2437. [Google Scholar] [CrossRef] [PubMed]
- Cañas, M.A.; Fábrega, M.J.; Giménez, R.; Badia, J.; Baldomà, L. Outer Membrane Vesicles from Probiotic and Commensal Escherichia Coli Activate NOD1-Mediated Immune Responses in Intestinal Epithelial Cells. Front. Microbiol. 2018, 9, 948. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garrido, N.; Fábrega, M.-J.; Vera, R.; Giménez, R.; Badia, J.; Baldomà, L. Membrane Vesicles from the Probiotic Nissle 1917 and Gut Resident Escherichia Coli Strains Distinctly Modulate Human Dendritic Cells and Subsequent T Cell Responses. J. Funct. Foods 2019, 61, 103495. [Google Scholar] [CrossRef]
- Diaz-Garrido, N.; Badia, J.; Baldomà, L. Modulation of Dendritic Cells by Microbiota Extracellular Vesicles Influences the Cytokine Profile and Exosome Cargo. Nutrients 2022, 14, 344. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Garrido, N.; Bonnin, S.; Riera, M.; Gíménez, R.; Badia, J.; Baldomà, L. Transcriptomic MicroRNA Profiling of Dendritic Cells in Response to Gut Microbiota-Secreted Vesicles. Cells 2020, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Fábrega, M.-J.; Rodríguez-Nogales, A.; Garrido-Mesa, J.; Algieri, F.; Badía, J.; Giménez, R.; Gálvez, J.; Baldomà, L. Intestinal Anti-Inflammatory Effects of Outer Membrane Vesicles from Escherichia Coli Nissle 1917 in DSS-Experimental Colitis in Mice. Front. Microbiol. 2017, 8, 1274. [Google Scholar] [CrossRef] [PubMed]
- Mayorgas, A.; Dotti, I.; Salas, A. Microbial Metabolites, Postbiotics, and Intestinal Epithelial Function. Mol. Nutr. Food Res. 2021, 65, 188. [Google Scholar] [CrossRef] [PubMed]
- Kavita; Om, H.; Chand, U.; Kushawaha, P.K. Postbiotics: An Alternative and Innovative Intervention for the Therapy of Inflammatory Bowel Disease. Microbiol. Res. 2024, 279, 127550. [Google Scholar] [CrossRef] [PubMed]
- Dosh, R.H.; Jordan-Mahy, N.; Sammon, C.; Le Maitre, C. Interleukin 1 Is a Key Driver of Inflammatory Bowel Disease-Demonstration in a Murine IL-1Ra Knockout Model. Oncotarget 2019, 10, 3559–3575. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Guo, S.; Ye, D.; Dokladny, K.; Alhmoud, T.; Ereifej, L.; Said, H.M.; Ma, T.Y. Mechanism of IL-1β Modulation of Intestinal Epithelial Barrier Involves P38 Kinase and Activating Transcription Factor-2 Activation. J. Immunol. 2013, 190, 6596–6606. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.J.; Mao, W.M.; Wang, Q.; Yang, G.G.; Wu, W.J.; Shao, S.X. MicroRNA-24 Inhibits Serotonin Reuptake Transporter Expression and Aggravates Irritable Bowel Syndrome. Biochem. Biophys. Res. Commun. 2016, 469, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Huang, Y.; Zhang, C.; Zhu, S.; Li, P.; Chen, X.; Hou, Z.; Liu, F. MicroRNA-200a Targets Cannabinoid Receptor 1 and Serotonin Transporter to Increase Visceral Hyperalgesia in Diarrhea-Predominant Irritable Bowel Syndrome Rats. J. Neurogastroenterol. Motil. 2018, 24, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, L.W.; Al-Sadi, R.; Ma, T.Y. IL-1β and the Intestinal Epithelial Tight Junction Barrier. Front. Immunol. 2021, 12, 767456. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.J.; Jonkers, D.M.; van Eijk, H.M.; Lindsey, P.J.; Dekker, J.; Buurman, W.A.; Masclee, A.A.M. Serotonergic Reinforcement of Intestinal Barrier Function Is Impaired in Irritable Bowel Syndrome. Aliment. Pharmacol. Ther. 2014, 40, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Saeid Seyedian, S.; Nokhostin, F.; Dargahi Malamir, M. A Review of the Diagnosis, Prevention, and Treatment Methods of Inflammatory Bowel Disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed]
- Annese, V. Genetics and Epigenetics of IBD. Pharmacol. Res. 2020, 159, 4892. [Google Scholar] [CrossRef] [PubMed]
- Dowdell, A.S.; Colgan, S.P. Metabolic Host–Microbiota Interactions in Autophagy and the Pathogenesis of Inflammatory Bowel Disease (Ibd). Pharmaceuticals 2021, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Haneishi, Y.; Furuya, Y.; Hasegawa, M.; Picarelli, A.; Rossi, M.; Miyamoto, J. Inflammatory Bowel Diseases and Gut Microbiota. Int. J. Mol. Sci. 2023, 24, 3817. [Google Scholar] [CrossRef] [PubMed]
- Losurdo, G.; Iannone, A.; Contaldo, A.; Ierardi, E.; Di Leo, A.; Principi, M. Escherichia Coli Nissle 1917 in Ulcerative Colitis Treatment: Systematic Review and Meta-Analysis. J. Gastrointest. Liver Dis. 2015, 24, 499–505. [Google Scholar] [CrossRef]
- Scaldaferri, F.; Gerardi, V.; Mangiola, F.; Lopetuso, L.R.; Pizzoferrato, M.; Petito, V.; Papa, A.; Stojanovic, J.; Poscia, A.; Cammarota, G.; et al. Role and Mechanisms of Action of Escherichia Coli Nissle 1917 in the Maintenance of Remission in Ulcerative Colitis Patients: An Update. World J. Gastroenterol. 2016, 22, 5505–5511. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut Microbiota-Derived Metabolites as Key Actors in Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Aggeletopoulou, I.; Kalafateli, M.; Tsounis, E.P.; Triantos, C. Exploring the Role of IL-1β in Inflammatory Bowel Disease Pathogenesis. Front. Med. 2024, 11, 7394. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Pi, X.; Jiang, Y.; Ren, G.; Liu, Z.; Liu, H.; Wang, M.; Sun, W.; Li, S.; Gao, Z.; et al. An Immuno-Blocking Agent Targeting IL-1β and IL-17A Reduces the Lesion of DSS-Induced Ulcerative Colitis in Mice. Inflammation 2021, 44, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal Bacteria-Dependent Indole Production Enhances Epithelial Barrier Function in the Colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef]
- Baudry, A.; Pietri, M.; Launay, J.-M.; Kellermann, O.; Schneider, B. Multifaceted Regulations of the Serotonin Transporter: Impact on Antidepressant Response. Front. Neurosci. 2019, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Woulfe, D.; Kilic, F. Post-Translational Modifications of Serotonin Transporter. Pharmacol. Res. 2019, 140, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Khalil, E.H.; Shaker, O.G.; Hasona, N.A. LncRNA H-19 and MiR-200a Implication and Frequency of LncRNA H-19 Rs2170425 SNP in Ulcerative Colitis and Crohn’s Disease. Comp. Clin. Path 2023, 32, 565–571. [Google Scholar] [CrossRef]
- Latorre, E.; Matheus, N.; Layunta, E.; Alcalde, A.I.; Mesonero, J.E. IL-10 Counteracts Proinflammatory Mediator Evoked Oxidative Stress in Caco-2 Cells. Mediat. Inflamm. 2014, 2014, 982639. [Google Scholar] [CrossRef] [PubMed]
- Frolova, L.; Drastich, P.; Rossmann, P.; Klimesova, K.; Tlaskalova-Hogenova, H. Expression of Toll-like Receptor 2 (TLR2), TLR4, and CD14 in Biopsy Samples of Patients with Inflammatory Bowel Diseases: Upregulated Expression of TLR2 in Terminal Ileum of Patients with Ulcerative Colitis. J. Histochem. Cytochem. 2008, 56, 267–274. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxid. Med. Cell. Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef] [PubMed]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight Junctions in Inflammatory Bowel Diseases and Inflammatory Bowel Disease Associated Colorectal Cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Nakai, D.; Miyake, M. Intestinal Membrane Function in Inflammatory Bowel Disease. Pharmaceutics 2023, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Ishihara, S. Enteric Microbiota-Mediated Serotonergic Signaling in Pathogenesis of Irritable Bowel Syndrome. Int. J. Mol. Sci. 2021, 22, 235. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.S.; Giménez, R.; Cañas, M.A.; Vera, R.; Díaz-Garrido, N.; Badia, J.; Baldomà, L. Extracellular Vesicles and Soluble Factors Secreted by Escherichia Coli Nissle 1917 and ECOR63 Protect against Enteropathogenic E. Coli-Induced Intestinal Epithelial Barrier Dysfunction. BMC Microbiol. 2019, 19, 166. [Google Scholar] [CrossRef]
- Kotla, N.G.; Rochev, Y. IBD Disease-Modifying Therapies: Insights from Emerging Therapeutics. Trends Mol. Med. 2023, 29, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Martyniak, A.; Medyńska-Przęczek, A.; Wędrychowicz, A.; Skoczeń, S.; Tomasik, P.J. Prebiotics, Probiotics, Synbiotics, Paraprobiotics and Postbiotic Compounds in IBD. Biomolecules 2021, 11, 1903. [Google Scholar] [CrossRef] [PubMed]
- Pesce, M.; Seguella, L.; Del Re, A.; Lu, J.; Palenca, I.; Corpetti, C.; Rurgo, S.; Sanseverino, W.; Sarnelli, G.; Esposito, G. Next-Generation Probiotics for Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 5466. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shin, T.-S.; Kim, J.S.; Jee, Y.-K.; Kim, Y.-K. A New Horizon of Precision Medicine: Combination of the Microbiome and Extracellular Vesicles. Exp. Mol. Med. 2022, 54, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Krzyżek, P.; Marinacci, B.; Vitale, I.; Grande, R. Extracellular Vesicles of Probiotics: Shedding Light on the Biological Activity and Future Applications. Pharmaceutics 2023, 15, 522. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ruiz, S.; Olivo-Martínez, Y.; Cordero, C.; Rodríguez-Lagunas, M.J.; Pérez-Cano, F.J.; Badia, J.; Baldoma, L. Microbiota-Derived Extracellular Vesicles as a Postbiotic Strategy to Alleviate Diarrhea and Enhance Immunity in Rotavirus-Infected Neonatal Rats. Int. J. Mol. Sci. 2024, 25, 1184. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Selander, R.K. Standard Reference Strains of Escherichia Coli from Natural Populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Arzt, M.; Deschamps, J.; Schmied, C.; Pietzsch, T.; Schmidt, D.; Tomancak, P.; Haase, R.; Jug, F. LABKIT: Labeling and Segmentation Toolkit for Big Image Data. Front. Comput. Sci. 2022, 4, 777728. [Google Scholar] [CrossRef]
- Huang, L.-K.; Wang, M.-J.J. Image Thresholding by Minimizing the Measures of Fuzziness. Pattern Recognit. 1995, 28, 41–51. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivo-Martínez, Y.; Martínez-Ruiz, S.; Cordero-Alday, C.; Bosch, M.; Badia, J.; Baldoma, L. Modulation of Serotonin-Related Genes by Extracellular Vesicles of the Probiotic Escherichia coli Nissle 1917 in the Interleukin-1β-Induced Inflammation Model of Intestinal Epithelial Cells. Int. J. Mol. Sci. 2024, 25, 5338. https://doi.org/10.3390/ijms25105338
Olivo-Martínez Y, Martínez-Ruiz S, Cordero-Alday C, Bosch M, Badia J, Baldoma L. Modulation of Serotonin-Related Genes by Extracellular Vesicles of the Probiotic Escherichia coli Nissle 1917 in the Interleukin-1β-Induced Inflammation Model of Intestinal Epithelial Cells. International Journal of Molecular Sciences. 2024; 25(10):5338. https://doi.org/10.3390/ijms25105338
Chicago/Turabian StyleOlivo-Martínez, Yenifer, Sergio Martínez-Ruiz, Cecilia Cordero-Alday, Manel Bosch, Josefa Badia, and Laura Baldoma. 2024. "Modulation of Serotonin-Related Genes by Extracellular Vesicles of the Probiotic Escherichia coli Nissle 1917 in the Interleukin-1β-Induced Inflammation Model of Intestinal Epithelial Cells" International Journal of Molecular Sciences 25, no. 10: 5338. https://doi.org/10.3390/ijms25105338