
Deciphering the Genetic Links between Psychological Stress, Autophagy, and Dermatological Health: Insights from Bioinformatics, Single-Cell Analysis, and Machine Learning in Psoriasis and Anxiety Disorders
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
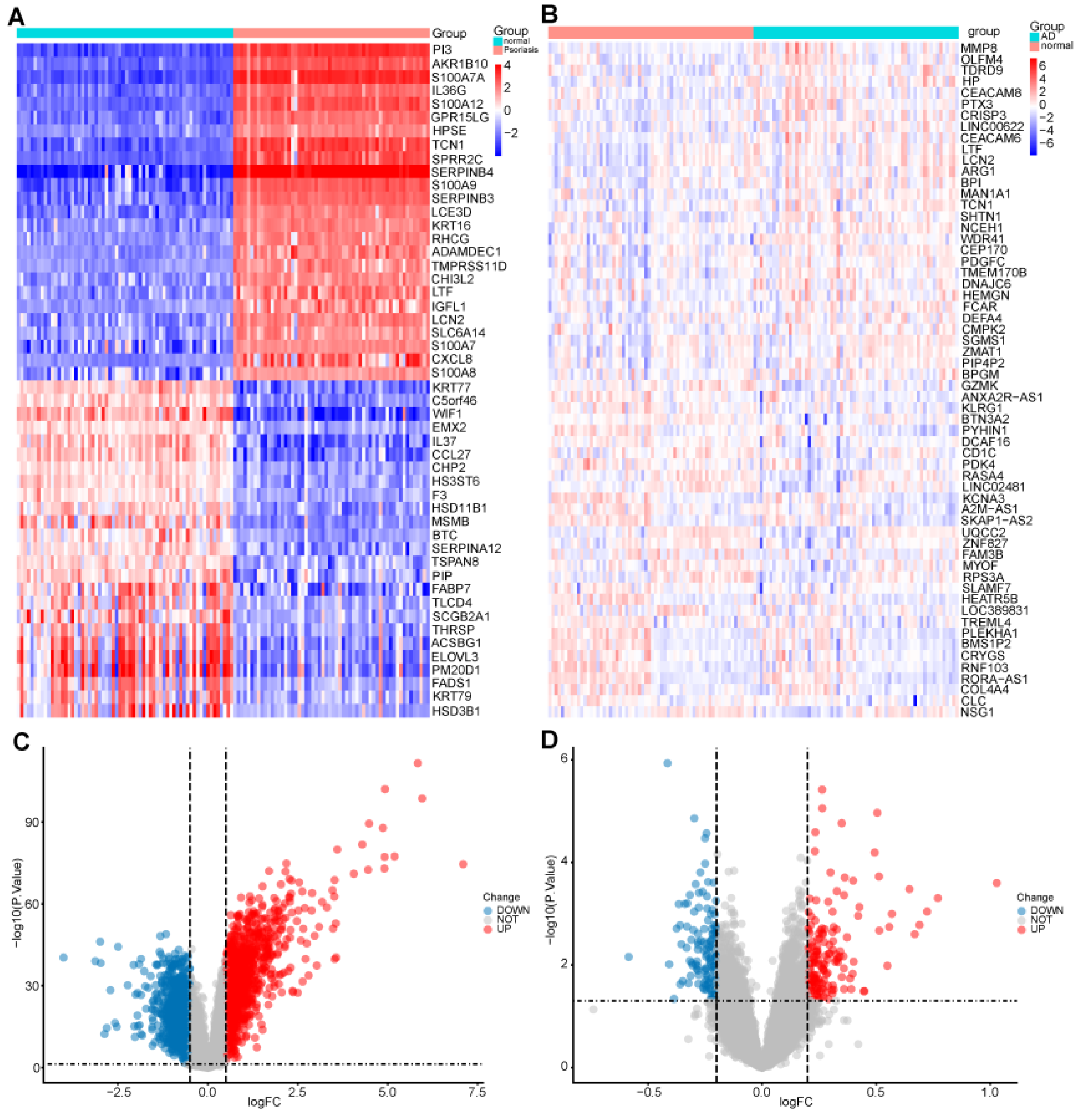
2. Results
2.1. Functional Enrichment Analysis
2.2. Hub Gene Identification
- The LASSO regression algorithm was applied to identify 13 potential candidate genes out of 16 candidate genes. (Figure 5A,B)
- The RF machine learning algorithm was also carried out to rank the 16 candidate genes in light of the variable importance of each gene, and the top 10 important genes with the MeanDecreaseGini score ranking were extracted. (Figure 5C)
- The SVM algorithm identified 14 genes with the lowest 5×CV error. (Figure 5D)
- We used the R package “Boruta”, and “mlbench” to run the BORUTA algorithm to rank the importance of the 16 candidate genes. The result showed that the whole 16 genes were confirmed to be retained without deduction. (Figure 5E,F)
- Then, we inputted the above 16 genes into the XGboosts algorithm classifier, and six genes were selected. (Figure 5G)
2.3. Diagnostic Model Construction
2.4. The Hub Genes and Immunocyte Infiltration in Different Disease Subtype Patterns
2.5. Biological Properties of Different Disease Subtype Patterns
2.6. Immunocyte Infiltration of the Disease Subtype
2.7. Transcription Factor and Hub Gene Interactions
2.8. Single-Cell Analysis Reveals Cell Subtypes
2.9. T-Cell Subtype Analysis
2.10. Pseudotime and Trajectory Analysis
- The CASP7 gene exhibited high expression in the initial phases of T-cell development, followed by a gradual decrease over time.
- The SLC7A5 gene displayed elevated expression in the early phases of T-cell development, followed by a decline, but interestingly, it resurged with significant expression in the later stages of cellular development.
- The ARG1 and YLPM1 genes showcased prominent expression in the advanced stages of T-cell development.
2.11. Cell Communication Analysis
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Processing
4.2. Screening for Autophagy-Related Genes
4.3. Identification of Differentially Expressed Genes (DEGs)
4.4. Identification of the Shared DEGs of Autophagy
4.5. Functional Enrichment Analysis
4.6. Gene Set Enrichment Analysis (GSEA)
4.7. Using Machine Learning Algorithms to Filter Core Hub Regulators
4.8. Consensus Clustering
4.9. Differences and Correlation Analysis of Immune Characteristics
4.10. Transcription Factor (TF)–Gene Interactions
4.11. Statistical Analysis
4.12. Single-Cell RNA-Sequencing Data
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Fang, Y.; Chen, H.; Zhang, T.; Yin, X.; Man, J.; Yang, L.; Lu, M. Global, Regional and National Burden of Anxiety Disorders from 1990 to 2019: Results from the Global Burden of Disease Study 2019. Epidemiol. Psychiatr. Sci. 2021, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, M.-K.; Hovatta, I. Genetic Insights into the Neurobiology of Anxiety. Trends Neurosci. 2023, 46, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Parisi, R.; Iskandar, I.Y.K.; Kontopantelis, E.; Augustin, M.; Griffiths, C.E.M.; Ashcroft, D.M. Global Psoriasis Atlas National, Regional, and Worldwide Epidemiology of Psoriasis: Systematic Analysis and Modelling Study. BMJ 2020, 369, m1590. [Google Scholar] [CrossRef] [PubMed]
- Jafferany, M.; Patel, A. Understanding Psychocutaneous Disease: Psychosocial & Psychoneuroimmunologic Perspectives. Int. J. Dermatol. 2020, 59, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Picardi, A.; Abeni, D. Stressful Life Events and Skin Diseases: Disentangling Evidence from Myth. Psychother. Psychosom. 2001, 70, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Pondeljak, N.; Lugović-Mihić, L. Stress-Induced Interaction of Skin Immune Cells, Hormones, and Neurotransmitters. Clin. Ther. 2020, 42, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Honeyman, J.F. Psychoneuroimmunology and the Skin. Acta Derm. Venereol. 2016, 96, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Puri, D.; Subramanyam, D. Stress—(Self) Eating: Epigenetic Regulation of Autophagy in Response to Psychological Stress. FEBS J. 2019, 286, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Zanetti, M.V.; Teixeira, A.L.; Uno, M.; Valiengo, L.L.; Soeiro-de-Souza, M.G.; Oba-Shinjo, S.M.; de Sousa, R.T.; Zarate, C.A.; Gattaz, W.F.; et al. Decreased AKT1/mTOR Pathway mRNA Expression in Short-Term Bipolar Disorder. Eur. Neuropsychopharmacol. 2015, 25, 468–473. [Google Scholar] [CrossRef]
- Tang, M.; Liu, T.; Jiang, P.; Dang, R. The Interaction between Autophagy and Neuroinflammation in Major Depressive Disorder: From Pathophysiology to Therapeutic Implications. Pharmacol. Res. 2021, 168, 105586. [Google Scholar] [CrossRef]
- Kotajima-Murakami, H.; Kobayashi, T.; Kashii, H.; Sato, A.; Hagino, Y.; Tanaka, M.; Nishito, Y.; Takamatsu, Y.; Uchino, S.; Ikeda, K. Effects of Rapamycin on Social Interaction Deficits and Gene Expression in Mice Exposed to Valproic Acid in Utero|Molecular Brain. Available online: https://link.springer.com/article/10.1186/s13041-018-0423-2 (accessed on 25 March 2024).
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-Based Unconventional Secretion of HMGB1 by Keratinocytes Plays a Pivotal Role in Psoriatic Skin Inflammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef]
- Klapan, K.; Simon, D.; Karaulov, A.; Gomzikova, M.; Rizvanov, A.; Yousefi, S.; Simon, H.-U. Autophagy and Skin Diseases. Front. Pharmacol. 2022, 13, 844756. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.M.; Deckert, J. Genetics of Anxiety Disorders. Curr. Psychiatry Rep. 2019, 21, 16. [Google Scholar] [CrossRef] [PubMed]
- The Critical Relationship Between Anxiety and Depression|American Journal of Psychiatry. Available online: https://ajp.psychiatryonline.org/doi/full/10.1176/appi.ajp.2020.20030305 (accessed on 3 May 2024).
- Jing, D.; Xiao, H.; Shen, M.; Chen, X.; Han, X.; Kuang, Y.; Zhu, W.; Xiao, Y. Association of Psoriasis with Anxiety and Depression: A Case–Control Study in Chinese Patients. Front. Med. 2021, 8, 771645. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, B.R.; Pio-Abreu, J.L.; Figueiredo, A.; Misery, L. Pruritus, Allergy and Autoimmunity: Paving the Way for an Integrated Understanding of Psychodermatological Diseases? Front. Allergy 2021, 2, 688999. [Google Scholar] [CrossRef]
- Wang, F.F.; Wang, Y.; Wang, L.; Wang, T.S.; Bai, Y.P. TIGIT Expression Levels on CD4+ T Cells Are Correlated with Disease Severity in Patients with Psoriasis. Clin. Exp. Dermatol. 2018, 43, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wang, Y.; Li, P.; Zhao, J. Gamma Delta T Cells and Their Pathogenic Role in Psoriasis. Front. Immunol. 2021, 12, 627139. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-N.; Dong, J.; Zhang, L.; Ouyang, D.; Cheng, Y.; Chen, A.F.; Lu, A.-P.; Cao, D.-S. HAMdb: A Database of Human Autophagy Modulators with Specific Pathway and Disease Information. J. Cheminform. 2018, 10, 34. [Google Scholar] [CrossRef]
- HADb: Human Autophagy Database. Available online: http://autophagy.lu/index.html (accessed on 20 September 2023).
- Diboun, I.; Wernisch, L.; Orengo, C.A.; Koltzenburg, M. Microarray Analysis after RNA Amplification Can Detect Pronounced Differences in Gene Expression Using Limma. BMC Genom. 2006, 7, 252. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- GSEABase. Available online: http://bioconductor.org/packages/GSEABase/ (accessed on 27 February 2024).
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Vasquez, M.M.; Hu, C.; Roe, D.J.; Chen, Z.; Halonen, M.; Guerra, S. Least Absolute Shrinkage and Selection Operator Type Methods for the Identification of Serum Biomarkers of Overweight and Obesity: Simulation and Application. BMC Med. Res. Methodol. 2016, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S. What Is a Support Vector Machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Mukherjee, D.P.; Das, P.; Gangopadhyay, A.; Chintha, A.R.; Kundu, S. Improved Random Forest for Classification. IEEE Trans. Image Process. 2018, 27, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; ACM: San Francisco, CA, USA, 2016; pp. 785–794. [Google Scholar]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Wolbers, M.; Koller, M.T.; Witteman, J.C.M.; Steyerberg, E.W. Prognostic Models with Competing Risks: Methods and Application to Coronary Risk Prediction. Epidemiology 2009, 20, 555–561. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An Interactive Venn Diagram Viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Steen, C.B.; Liu, C.L.; Alizadeh, A.A.; Newman, A.M. Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 2117, pp. 135–157. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 1711, pp. 243–259. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Kim, H.J.; Kameyama, N.; Nazarian, R.; Der, E.; Cohen, S.; Guttman-Yassky, E.; Putterman, C.; Krueger, J.G. Single-Cell Transcriptomics Applied to Emigrating Cells from Psoriasis Elucidate Pathogenic versus Regulatory Immune Cell Subsets. J. Allergy Clin. Immunol. 2021, 148, 1281–1292. [Google Scholar] [CrossRef]
- Ma, F.; Plazyo, O.; Billi, A.C.; Tsoi, L.C.; Xing, X.; Wasikowski, R.; Gharaee-Kermani, M.; Hile, G.; Jiang, Y.; Harms, P.W.; et al. Single Cell and Spatial Sequencing Define Processes by Which Keratinocytes and Fibroblasts Amplify Inflammatory Responses in Psoriasis. Nat. Commun. 2023, 14, 3455. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed Graph Embedding Resolves Complex Single-Cell Trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.-L.; Chang, L.-S. Deciphering the Genetic Links between Psychological Stress, Autophagy, and Dermatological Health: Insights from Bioinformatics, Single-Cell Analysis, and Machine Learning in Psoriasis and Anxiety Disorders. Int. J. Mol. Sci. 2024, 25, 5387. https://doi.org/10.3390/ijms25105387
Liu X-L, Chang L-S. Deciphering the Genetic Links between Psychological Stress, Autophagy, and Dermatological Health: Insights from Bioinformatics, Single-Cell Analysis, and Machine Learning in Psoriasis and Anxiety Disorders. International Journal of Molecular Sciences. 2024; 25(10):5387. https://doi.org/10.3390/ijms25105387
Chicago/Turabian StyleLiu, Xiao-Ling, and Long-Sen Chang. 2024. "Deciphering the Genetic Links between Psychological Stress, Autophagy, and Dermatological Health: Insights from Bioinformatics, Single-Cell Analysis, and Machine Learning in Psoriasis and Anxiety Disorders" International Journal of Molecular Sciences 25, no. 10: 5387. https://doi.org/10.3390/ijms25105387