
Kinase Inhibitors and Kinase-Targeted Cancer Therapies: Recent Advances and Future Perspectives

Abstract
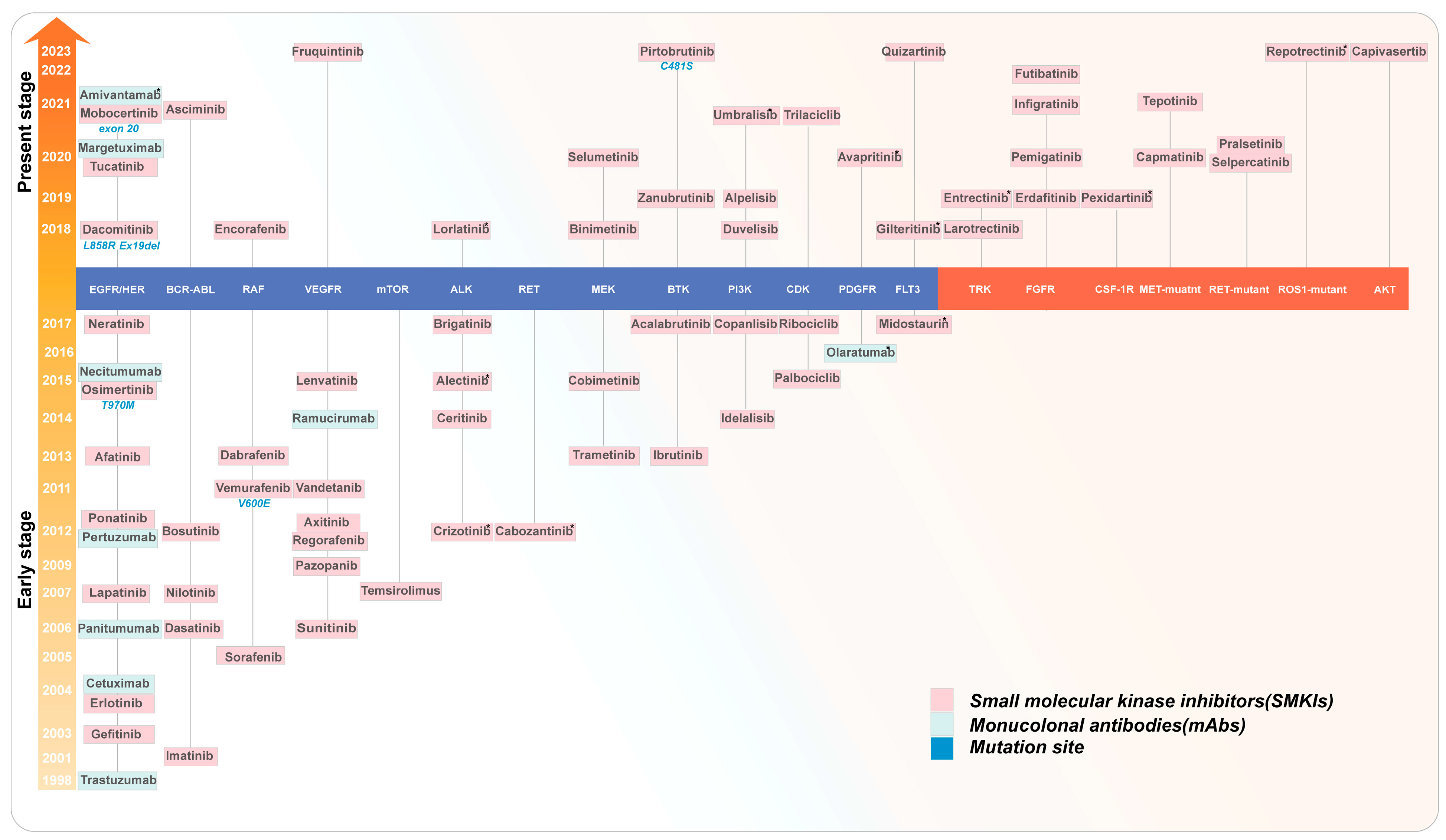
:1. Introduction
2. Types of Kinase Inhibitors
2.1. Mechanism of Action and Classification of SMKIs
2.1.1. Structural Features of Kinase
2.1.2. ATP-Competitive Inhibitors
2.1.3. Allosteric Inhibitors
2.1.4. Covalent Inhibitors
2.1.5. Type Selection for the Development of SMKIs

{kind=link}
{kind=link}
{kind=link}
| Compound Name | First Approved | Target | Type | Reference |
|---|---|---|---|---|
| Quizartinib | 2023 | FLT3 inhibitors | Type II | [58] |
| Capivasertib | 2023 | AKT-1, AKT-2, AKT-3 | Type I | [59] |
| Pirtobrutinib | 2023 | BTK C481S inhibitor | Type II | [39] |
| Repotrectinib | 2023 | ALK, ROS1, TRKA, TRKC | Type II | [40] |
| Fruquintinib | 2023 | VEGFR1, VEGFR2, VEGFR3 | Type I/II | [60] |
| Futibatinib | 2022 | FGFRs | Type V | [45] |
| Tepotinib | 2021 | c-Met | Type I | [61] |
| Amivantamab | 2021 | EGFR, c-Met | [62] | |
| Asciminib | 2021 | BCR–ABL | Type IV | [42] |
| Infigratinib | 2021 | FGFR1, FGFR2, FGFR3, FGFR4 | Type I/II | [63] |
| Mobocertinib | 2021 | EGFR exon 20, HER2 exon 20 | Type V | [46] |
| Umbralisib | 2021 | CK1ε, PI3Kδ | Type I/II | [64] |
| Tucatinib | 2020 | HER2 | Type I/II | [65] |
| Avapritinib | 2020 | PDGFRα, c-Kit | Type I | [66] |
| Capmatinib | 2020 | c-Met | Type I | [32] |
| Margetuximab | 2020 | HER2 | Monoclonal antibody | [67] |
| Pemigatinib | 2020 | FGFR1, FGFR2, FGFR3 | Type I/II | [68] |
| Pralsetinib | 2020 | RET | Type I | [69] |
| Selpercatinib | 2020 | RET | Type I | [33] |
| Selumetinib | 2020 | MEK1, MEK2 | Type III | [44] |
| Ripretinib | 2020 | EGFR, PDGFRα, c-Kit | TypeII | [57] |
| Alpelisib | 2019 | PI3Kα | Type I/II | [70] |
| Entrectinib | 2019 | ALK, ROS1, TRKA, TRKB, TRKC | Type I | [34] |
| Erdafitinib | 2019 | FGFR1, FGFR2, FGFR3, FGFR4 | Type I | [71] |
| Pexidartinib | 2019 | CSF-1R, FLT3, c-Kit | Type III | [72] |
| Zanubrutinib | 2019 | BTK | Type V | [47] |
| Duvelisib | 2018 | PI3Kγ, PI3Kδ | Type I/II | [73] |
| Encorafenib | 2018 | BRAF | Type I/II | [74] |
| Gilteritinib | 2018 | AXL, FLT3 | Type I | [36] |
| Dacomitinib | 2018 | EGFR, EGFRL858R, EGFR-Ex19del, HER2, HER4 | Type V | [48] |
| Lorlatinib | 2018 | ALK, ROS1 | Type I/II | [75] |
| Binimetinib | 2018 | MEK1, MEK2 | TypeIII/IV | [76] |
| Larotrectinib | 2018 | TRKA, TRKB, TRKC | Type I/II | [77] |
2.2. Monoclonal Antibodies
3. FDA Approved Kinase Inhibitors in Oncology in 2018–2024
3.1. Tropomyosin Receptor Kinase (TRK)
3.2. Fibroblast Growth Factor Receptors (FGFRs)
3.3. Colony-Stimulating Factor 1 Receptor (CSF-1R)
3.4. Cellular Mesenchymal-to-Epithelial Transition Factor (c-MET)
3.5. Proto-Oncogene Tyrosine–Protein Kinase Receptor (RET)
3.6. ROS Proto-Oncogene 1 (ROS1)
3.7. Protein Kinase B (AKT)
4. New Strategies for Kinase-Targeted Cancer Therapies
4.1. Monoclonal Antibodies and Derivatives
4.1.1. Monoclonal Antibodies
4.1.2. Nanobodies
4.2. Kinase Degraders
4.2.1. PROTACs
4.2.2. Molecular Glue Degraders
4.2.3. Other Degraders
4.3. Protein–Kinase Interaction Inhibitors (PKIIs)
5. Future Perspectives
5.1. Potential New Targets
5.2. Computing to Accelerate Drug Development and Repurposing
5.3. Overcoming Drug Resistance
5.4. Crossing the Blood–Brain Barrier
5.5. Combination with Immunotherapy
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AbTACs | Antibody-based PROTACs |
| AURKs | Aurora kinases |
| AUTOTAC | Autophagy-targeting chimera |
| BBB | Blood–brain barrier |
| BTK | Bruton tyrosine kinase |
| c-MET | Cellular mesenchymal-to-epithelial transition factor |
| CNS | Central nervous system |
| CRBN | Cereblon |
| CLL | Chronic lymphocytic leukemia |
| CSF-1R | Colony-stimulating factor 1 receptor |
| CR | Complete response |
| dTAGs | Degradation tags |
| DC | Dendritic cell |
| DOR | Duration of response |
| EFS | Event-free survival |
| EGFR | Epidermal growth factor receptor |
| eTPD | Extracellular targeted protein degradation |
| FGFRs | Fibroblast growth factor receptors |
| HyT-PD | Hydrophobic tagging-based protein degradation |
| ICIs | Immune checkpoint inhibitors |
| IGF-1R | Insulin-like growth factor 1 receptor |
| IND | Investigational New Drug |
| LYTACs | Lysosome-targeting chimeras |
| LTR | Lysosome-targeting receptor |
| mAbs | Monoclonal antibodies |
| NMPA | National Medical Products Administration |
| NEK7 | NIMA-related kinase 7 |
| ORR | Overall response rate |
| AKT | Protein kinase B |
| POI | Protein of interest |
| PKIIs | Protein–kinase interaction inhibitors |
| PPI | Protein–protein interaction |
| PPIs | Protein–protein interactions |
| PROTACs | Proteolysis-targeting chimeras |
| RET | Proto-oncogene tyrosine protein kinase receptor |
| ROR1 | Receptor tyrosine kinase-like orphan receptor 1 |
| RTKs | Receptor tyrosine kinases |
| ROS1 | ROS proto-oncogene 1 |
| SMKIs | Small-molecule kinase inhibitors |
| TPD | Targeted protein degradation |
| TRK | Tropomyosin receptor kinase |
| TME | Tumor microenvironment |
| TAMs | Tumor-associated macrophages |
| FDA | U.S. Food and Drug Administration |
| UC | Urothelial carcinoma |
References
- Kanev, G.K.; de Graaf, C.; de Esch, I.J.P.; Leurs, R.; Würdinger, T.; Westerman, B.A.; Kooistra, A.J. The Landscape of Atypical and Eukaryotic Protein Kinases. Trends Pharmacol. Sci. 2019, 40, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, X.; Tang, M.; Zhong, G.; Si, Y.; Li, H.; Zhu, F.; Liao, Q.; Li, L.; Zhao, J.; et al. A Subcellular Map of the Human Kinome. eLife 2021, 10, e64943. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Schork, N.J. Distribution Analysis of Nonsynonymous Polymorphisms within the Human Kinase Gene Family. Genomics 2007, 90, 49–58. [Google Scholar] [CrossRef]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase Mutations in Human Disease: Interpreting Genotype–Phenotype Relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Author Correction: Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discov. 2021, 20, 798. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Dewanjee, S.; Li, Y.; Jha, N.K.; Chen, Z.-S.; Kumar, A.; Vishakha; Behl, T.; Jha, S.K.; Tang, H. Advancements in Clinical Aspects of Targeted Therapy and Immunotherapy in Breast Cancer. Mol. Cancer 2023, 22, 105. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Fu, M.; Hu, Y.; Wei, Y.; Wei, X.; Luo, M. Regulation and Signaling Pathways in Cancer Stem Cells: Implications for Targeted Therapy for Cancer. Mol. Cancer 2023, 22, 172. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, Z. Non-Small Cell Lung Cancer Targeted Therapy: Drugs and Mechanisms of Drug Resistance. Int. J. Mol. Sci. 2022, 23, 15056. [Google Scholar] [CrossRef] [PubMed]
- Bansal, I.; Pandey, A.K.; Ruwali, M. Small-Molecule Inhibitors of Kinases in Breast Cancer Therapy: Recent Advances, Opportunities, and Challenges. Front. Pharmacol. 2023, 14, 1244597. [Google Scholar] [CrossRef] [PubMed]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural Features of the Protein Kinase Domain and Targeted Binding by Small-Molecule Inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef] [PubMed]
- FDA. Drugs@FDA: NDA 021335 (Imatinib Mesylate) ORIG-1 Label. 2001. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/021588lbl.pdf (accessed on 3 March 2024).
- Scott, E.C.; Baines, A.C.; Gong, Y.; Moore, R.; Pamuk, G.E.; Saber, H.; Subedee, A.; Thompson, M.D.; Xiao, W.; Pazdur, R.; et al. Trends in the Approval of Cancer Therapies by the FDA in the Twenty-First Century. Nat. Rev. Drug Discov. 2023, 22, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Sequist, L.V.; Lin, J.J. Third-Generation EGFR and ALK Inhibitors: Mechanisms of Resistance and Management. Nat. Rev. Clin. Oncol. 2022, 19, 499–514. [Google Scholar] [CrossRef] [PubMed]
- FDA. Drugs@FDA: BLA 103792 (Trastuzumab) ORIG-1 Label. 1998. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/1998/trasgen092598lb.pdf (accessed on 3 March 2024).
- Abdolvahab, M.H.; Karimi, P.; Mohajeri, N.; Abedini, M.; Zare, H. Targeted Drug Delivery Using Nanobodies to Deliver Effective Molecules to Breast Cancer Cells: The Most Attractive Application of Nanobodies. Cancer Cell Int. 2024, 24, 67. [Google Scholar] [CrossRef] [PubMed]
- Klein, M. Targeting Protein-Protein Interactions to Inhibit Cyclin-Dependent Kinases. Pharmaceuticals 2023, 16, 519. [Google Scholar] [CrossRef] [PubMed]
- Zerihun, M.; Rubin, S.J.S.; Silnitsky, S.; Qvit, N. An Update on Protein Kinases as Therapeutic Targets—Part II: Peptides as Allosteric Protein Kinase C Modulators Targeting Protein–Protein Interactions. Int. J. Mol. Sci. 2023, 24, 17504. [Google Scholar] [CrossRef] [PubMed]
- Blaquier, J.B.; Ortiz-Cuaran, S.; Ricciuti, B.; Mezquita, L.; Cardona, A.F.; Recondo, G. Tackling Osimertinib Resistance in EGFR-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 3579–3591. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Moccia, M.; Briggs, D.C.; Bharate, J.B.; Lakkaniga, N.R.; Knowles, P.; Yan, W.; Tran, P.; Kharbanda, A.; Wang, X.; et al. Discovery of N-Trisubstituted Pyrimidine Derivatives as Type I RET and RET Gatekeeper Mutant Inhibitors with a Novel Kinase Binding Pose. J. Med. Chem. 2022, 65, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F. Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases. Sci. Pharm. 2023, 91, 18. [Google Scholar] [CrossRef]
- McCoull, W.; Boyd, S.; Brown, M.R.; Coen, M.; Collingwood, O.; Davies, N.L.; Doherty, A.; Fairley, G.; Goldberg, K.; Hardaker, E.; et al. Optimization of an Imidazo[1,2-a]Pyridine Series to Afford Highly Selective Type I1/2 Dual Mer/Axl Kinase Inhibitors with In Vivo Efficacy. J. Med. Chem. 2021, 64, 13524–13539. [Google Scholar] [CrossRef] [PubMed]
- Akwata, D.; Kempen, A.L.; Lamptey, J.; Dayal, N.; Brauer, N.R.; Sintim, H.O. Discovery of Imidazo[1,2-b]Pyridazine-Containing TAK1 Kinase Inhibitors with Excellent Activities against Multiple Myeloma. RSC Med. Chem. 2024, 15, 178–192. [Google Scholar] [CrossRef]
- El-Wakil, M.H.; Teleb, M. Transforming Type II to Type I C-Met Kinase Inhibitors via Combined Scaffold Hopping and Structure-Guided Synthesis of New Series of 1,3,4-Thiadiazolo[2,3-c]-1,2,4-Triazin-4-One Derivatives. Bioorg Chem. 2021, 116, 105304. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase Hinge Binding Scaffolds and Their Hydrogen Bond Patterns. Bioorg Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2024 Update. Pharmacol. Res. 2024, 200, 107059. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Smaill, J.B.; Ding, K. New Promise and Opportunities for Allosteric Kinase Inhibitors. Angew. Chem. Int. Ed. 2020, 59, 13764–13776. [Google Scholar] [CrossRef] [PubMed]
- Laufkötter, O.; Hu, H.; Miljković, F.; Bajorath, J. Structure- and Similarity-Based Survey of Allosteric Kinase Inhibitors, Activators, and Closely Related Compounds. J. Med. Chem. 2022, 65, 922–934. [Google Scholar] [CrossRef] [PubMed]
- TRUQAPTM (Capivasertib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/218197s000lbl.pdf (accessed on 7 March 2024).
- Dhillon, S. Capmatinib: First Approval. Drugs 2020, 80, 1125–1131. [Google Scholar] [CrossRef]
- Markham, A. Selpercatinib: First Approval. Drugs 2020, 80, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Al-Salama, Z.T.; Keam, S.J. Entrectinib: First Global Approval. Drugs 2019, 79, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Erdafitinib: First Global Approval. Drugs 2019, 79, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Zhang, C.; Lin, K.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and Overriding the Structural Mechanism of the Quizartinib-Resistant FLT3 “Gatekeeper” F691L Mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef] [PubMed]
- De, S.K. Pirtobrutinib: First Non-Covalent Tyrosine Kinase Inhibitor for Treating Relapsed or Refractory Mantle Cell Lymphoma in Adults. Curr. Med. Chem. 2023, 31. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Repotrectinib: First Approval. Drugs 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Encorafenib and Binimetinib: First Global Approvals. Drugs 2018, 78, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Asciminib: First Approval. Drugs 2022, 82, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Keam, S.J. Selumetinib: First Approval. Drugs 2020, 80, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Futibatinib: First Approval. Drugs 2022, 82, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Mobocertinib: First Approval. Drugs 2021, 81, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Zanubrutinib: First Approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef]
- Dungo, R.T.; Keating, G.M. Afatinib: First Global Approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Kharkar, P.S. Covalent Inhibitors: An Ambitious Approach for the Discovery of Newer Oncotherapeutics. Drug Dev. Res. 2024, 85, e22132. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, D.; Cheng, X. Recent Advances in Covalent Drug Discovery. Pharmaceuticals 2023, 16, 663. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Faridoon; Ng, R.; Zhang, G.; Li, J.J. An Update on the Discovery and Development of Reversible Covalent Inhibitors. Med. Chem. Res. 2023, 32, 1039–1062. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Efraim, M.; Mayer, J.; Trněný, M.; McDonald, V.; Bird, R.; Regenbogen, T.; Garg, M.; Kaplan, Z.; Tzvetkov, N.; et al. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. N. Engl. J. Med. 2022, 386, 1421–1431. [Google Scholar] [CrossRef]
- Ucpinar, S.; Smith, P.F.; Long, L.; Li, F.; Yan, H.; Wadhwa, J.; Chu, K.A.; Shu, J.; Nunn, P.; Li, M. Rilzabrutinib, a Reversible Covalent Bruton’s Tyrosine Kinase Inhibitor: Absorption, Metabolism, Excretion, and Absolute Bioavailability in Healthy Participants. Clin. Transl. Sci. 2023, 16, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.B.; Ebata, K.; Randeria, H.S.; Rosendahl, M.S.; Cedervall, E.P.; Morales, T.H.; Hanson, L.M.; Brown, N.E.; Gong, X.; Stephens, J.R.; et al. Pirtobrutinib Preclinical Characterization: A Highly Selective, Non-Covalent (Reversible) BTK Inhibitor. Blood 2023, 142, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef] [PubMed]
- Chao, Q.; Sprankle, K.G.; Grotzfeld, R.M.; Lai, A.G.; Carter, T.A.; Velasco, A.M.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; James, J.; et al. Identification of N-(5-Tert-Butyl-Isoxazol-3-Yl)-N′-{4-[7-(2-Morpholin-4-Yl-Ethoxy)Imidazo[2,1-b][1,3]Benzothiazol-2-Yl]Phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor. J. Med. Chem. 2009, 52, 7808–7816. [Google Scholar] [CrossRef] [PubMed]
- Addie, M.; Ballard, P.; Buttar, D.; Crafter, C.; Currie, G.; Davies, B.R.; Debreczeni, J.; Dry, H.; Dudley, P.; Greenwood, R.; et al. Discovery of 4-Amino-N-[(1 S)-1-(4-Chlorophenyl)-3-Hydroxypropyl]-1-(7 H-Pyrrolo[2,3-d]Pyrimidin-4-Yl)Piperidine-4-Carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. J. Med. Chem. 2013, 56, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Fruquintinib: First Global Approval. Drugs 2018, 78, 1757–1761. [Google Scholar] [CrossRef]
- Bladt, F.; Faden, B.; Friese-Hamim, M.; Knuehl, C.; Wilm, C.; Fittschen, C.; Grädler, U.; Meyring, M.; Dorsch, D.; Jaehrling, F.; et al. EMD 1214063 and EMD 1204831 Constitute a New Class of Potent and Highly Selective C-Met Inhibitors. Clin. Cancer Res. 2013, 19, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Amivantamab: First Approval. Drugs 2021, 81, 1349–1353. [Google Scholar] [CrossRef]
- Kang, C. Infigratinib: First Approval. Drugs 2021, 81, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.-X.; et al. Silencing C-Myc Translation as a Therapeutic Strategy through Targeting PI3Kδ and CK1ε in Hematological Malignancies. Blood 2017, 129, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, A. Tucatinib: First Approval. Drugs 2020, 80, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Avapritinib: First Approval. Drugs 2020, 80, 433–439. [Google Scholar] [CrossRef]
- Markham, A. Margetuximab: First Approval. Drugs 2021, 81, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Pralsetinib: First Approval. Drugs 2020, 80, 1865–1870. [Google Scholar] [CrossRef]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a Potent and Selective Phosphatidylinositol-3 Kinase Alpha Inhibitor Selected for Clinical Evaluation. Bioorg Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, M.; Debucquoy, A.; Gonnissen, A.; Dok, R.; Isebaert, S.; Devos, E.; McBride, W.; Haustermans, K. In Vitro and in Vivo Evaluation of the Radiosensitizing Effect of a Selective FGFR Inhibitor (JNJ-42756493) for Rectal Cancer. BMC Cancer 2015, 15, 946. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.G.; Faia, K.L.; DiNitto, J.P.; Ali, J.A.; White, K.F.; Brophy, E.E.; Pink, M.M.; Proctor, J.L.; Lussier, J.; Martin, C.M.; et al. PI3K-δ and PI3K-γ Inhibition by IPI-145 Abrogates Immune Responses and Suppresses Activity in Autoimmune and Inflammatory Disease Models. Chem. Biol. 2013, 20, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Hahn, E.; Chavira, R.; Wollenberg, L.; Tan, W.; Reddy, M.B. Impact of Posaconazole and Diltiazem on Pharmacokinetics of Encorafenib, a BRAF V600 Kinase Inhibitor for Melanoma and Colorectal Cancer with BRAF Mutations. Clin. Transl. Sci. 2023, 16, 2675–2686. [Google Scholar] [CrossRef]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.-L.; Dinh, D.; et al. Discovery of (10 R.)-7-Amino-12-Fluoro-2,10,16-Trimethyl-15-Oxo-10,15,16,17-Tetrahydro- 2H-8,4-(Metheno)Pyrazolo[4,3-h ][2,5,11]-Benzoxadiazacyclotetradecine-3-Carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-Ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; Van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for Patients with Advanced Melanoma Harbouring NRAS or Val600 BRAF Mutations: A Non-Randomised, Open-Label Phase 2 Study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An Oncogenic NTRK Fusion in a Soft Tissue Sarcoma Patient with Response to the Tropomyosin-Related Kinase (TRK) Inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef]
- Brazel, D.; Nagasaka, M. The Development of Amivantamab for the Treatment of Non-Small Cell Lung Cancer. Respir. Res. 2023, 24, 256. [Google Scholar] [CrossRef] [PubMed]
- RYBREVANTTM. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761210s000lbl.pdf (accessed on 8 March 2024).
- Latham, B.D.; Geffert, R.M.; Jackson, K.D. Kinase Inhibitors FDA-Approved 2018–2023: Drug Targets, Metabolic Pathways, and Drug-Induced Toxicities. Drug Metab. Dispos. 2024, 52, DMD-MR-2023-001430. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2020 Update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2022 Update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2023 Update. Pharmacol. Res. 2023, 187, 106552. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Larotrectinib: First Global Approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. New Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Kojadinovic, A.; Laderian, B.; Mundi, P.S. Targeting TRK: A Fast-Tracked Application of Precision Oncology and Future Directions. Crit. Rev. Oncol. Hematol. 2021, 165, 103451. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Lu, X. Selective Type II TRK Inhibitors Overcome xDFG Mutation Mediated Acquired Resistance to the Second-Generation Inhibitors Selitrectinib and Repotrectinib. Acta Pharm. Sin. B 2024, 14, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, J.; Yan, W.; Liu, Z.; Liu, S.; Fang, W. FGFR Families: Biological Functions and Therapeutic Interventions in Tumors. MedComm 2023, 4, e367. [Google Scholar] [CrossRef]
- Benjamin, D.J.; Hsu, R. Treatment Approaches for FGFR-Altered Urothelial Carcinoma: Targeted Therapies and Immunotherapy. Front. Immunol. 2023, 14, 1258388. [Google Scholar] [CrossRef] [PubMed]
- PEMAZYRETM (Pemigatinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213736s000lbl.pdf (accessed on 10 March 2024).
- TRUSELTIQ (Infigratinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214622s000lbl.pdf (accessed on 10 March 2024).
- LYTGOBI® (Futibatinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214801Orig1s000lbledt.pdf (accessed on 11 March 2024).
- Wu, Q.; Ellis, H.; Siravegna, G.; Michel, A.G.; Norden, B.L.; Fece De La Cruz, F.; Balasooriya, E.R.; Zhen, Y.; Silveira, V.S.; Che, J.; et al. Landscape of Clinical Resistance Mechanisms to FGFR Inhibitors in FGFR2-Altered Cholangiocarcinoma. Clin. Cancer Res. 2024, 30, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Loriot, Y.; Brandi, G.; Tavolari, S.; Wainberg, Z.A.; Katoh, M. FGFR-Targeted Therapeutics: Clinical Activity, Mechanisms of Resistance and New Directions. Nat. Rev. Clin. Oncol. 2024, 21, 312–329. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Kang, Y.-K.; Lee, K.-W.; Qin, S.; Yamaguchi, K.; Kim, I.-H.; Saeed, A.; Oh, S.C.; Li, J.; Turk, H.M.; et al. Bemarituzumab as First-Line Treatment for Locally Advanced or Metastatic Gastric/Gastroesophageal Junction Adenocarcinoma: Final Analysis of the Randomized Phase 2 FIGHT Trial. Gastric Cancer 2024, 27, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Kim, H.; Jun, J.; Kang, D.; Bae, H.; Cho, H.; Hah, J.-M. Discovery of N-(5-Amido-2-Methylphenyl)-5-Methylisoxazole-3-Carboxamide as Dual CSF-1R/c-Kit Inhibitors with Improved Stability and BBB Permeability. Eur. J. Med. Chem. 2024, 268, 116253. [Google Scholar] [CrossRef]
- Laoui, D.; Van Overmeire, E.; De Baetselier, P.; Van Ginderachter, J.A.; Raes, G. Functional Relationship between Tumor-Associated Macrophages and Macrophage Colony-Stimulating Factor as Contributors to Cancer Progression. Front. Immunol. 2014, 5, 113383. [Google Scholar] [CrossRef] [PubMed]
- TURALIOTM (Pexidartinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211810s000lbl.pdf (accessed on 13 March 2024).
- Voissière, A.; Gomez-Roca, C.; Chabaud, S.; Rodriguez, C.; Nkodia, A.; Berthet, J.; Montane, L.; Bidaux, A.-S.; Treilleux, I.; Eberst, L.; et al. The CSF-1R Inhibitor Pexidartinib Affects FLT3-Dependent DC Differentiation and May Antagonize Durvalumab Effect in Patients with Advanced Cancers. Sci. Transl. Med. 2024, 16, eadd1834. [Google Scholar] [CrossRef]
- Becker, A.M.D.; Decker, A.H.; Flórez-Grau, G.; Bakdash, G.; Röring, R.J.; Stelloo, S.; Vermeulen, M.; Piet, B.; Aarntzen, E.H.J.G.; Verdoes, M.; et al. Inhibition of CSF-1R and IL-6R Prevents Conversion of cDC2s into Immune Incompetent Tumor-Induced DC3s Boosting DC-Driven Therapy Potential. Cell Rep. Med. 2024, 5, 101386. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-W.; Hsiao, H.-W.; Chen, J.-P.; Tzeng, S.-F.; Tsai, C.-H.; Wu, C.-Y.; Hsieh, H.-H.; Carmona, S.J.; Andreatta, M.; Di Conza, G.; et al. A CSF-1R-Blocking Antibody/IL-10 Fusion Protein Increases Anti-Tumor Immunity by Effectuating Tumor-Resident CD8+ T Cells. Cell Rep. Med. 2023, 4, 101154. [Google Scholar] [CrossRef] [PubMed]
- Spitaleri, G.; Trillo Aliaga, P.; Attili, I.; Del Signore, E.; Corvaja, C.; Corti, C.; Uliano, J.; Passaro, A.; de Marinis, F. MET in Non-Small-Cell Lung Cancer (NSCLC): Cross ‘a Long and Winding Road’ Looking for a Target. Cancers 2023, 15, 4779. [Google Scholar] [CrossRef]
- TABRECTATM (Capmatinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213591s000lbl.pdf (accessed on 13 March 2024).
- TEPMETKO® (Tepotinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214096s000lbl.pdf (accessed on 14 March 2024).
- Hartmaier, R.J.; Markovets, A.A.; Ahn, M.J.; Sequist, L.V.; Han, J.-Y.; Cho, B.C.; Yu, H.A.; Kim, S.-W.; Yang, J.C.-H.; Lee, J.-S.; et al. Osimertinib + Savolitinib to Overcome Acquired MET-Mediated Resistance in Epidermal Growth Factor Receptor–Mutated, MET-Amplified Non–Small Cell Lung Cancer: TATTON. Cancer Discov. 2023, 13, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.A. Targeting the RET Tyrosine Kinase in Neuroblastoma: A Review and Application of a Novel Selective Drug Design Strategy. Biochem. Pharmacol. 2023, 216, 115751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X.; Li, Q.-Q.; Cai, J.; Wu, J.-Z.; Wang, J.-J.; Zhang, M.-Y.; Wang, Q.-X.; Tong, Z.-J.; Yang, J.; Wei, T.-H.; et al. Unraveling the Promise of RET Inhibitors in Precision Cancer Therapy by Targeting RET Mutations. J. Med. Chem. 2024, 67, 4346–4375. [Google Scholar] [CrossRef] [PubMed]
- GAVRETOTM (Pralsetinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213721s000lbl.pdf (accessed on 15 March 2024).
- RETEVMOTM (Selpercatinib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213246s000lbl.pdf (accessed on 15 March 2024).
- Zhou, C.; Solomon, B.; Loong, H.H.; Park, K.; Pérol, M.; Arriola, E.; Novello, S.; Han, B.; Zhou, J.; Ardizzoni, A.; et al. First-Line Selpercatinib or Chemotherapy and Pembrolizumab in RET Fusion–Positive NSCLC. N. Engl. J. Med. 2023, 389, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bradford, D.; Larkins, E.; Pai-Scherf, L.H.; Chatterjee, S.; Mishra-Kalyani, P.S.; Wearne, E.; Helms, W.S.; Ayyoub, A.; Bi, Y.; et al. FDA Approval Summary: Pralsetinib for the Treatment of Lung and Thyroid Cancers With RET Gene Mutations or Fusions. Clin. Cancer Res. 2021, 27, 5452–5456. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gouda, M.A.; Iorgulescu, J.B.; Dadu, R.; Patel, K.; Sherman, S.; Cabanillas, M.; Hu, M.; Castellanos, L.E.; Amini, B.; et al. Adaptive Darwinian Off-Target Resistance Mechanisms to Selective RET Inhibition in RET Driven Cancer. NPJ Precis. Oncol. 2024, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Khatri, U.; Shen, T.; Wu, J. Progress and Challenges in RET-Targeted Cancer Therapy. Front. Med. 2023, 17, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.R.; Nusser, K.; Jones, K.; Shinde, P.; Keddy, C.; Beach, C.Z.; Aguero, E.; Force, J.; Shinde, U.; Davare, M.A. Discovery of Oncogenic ROS1 Missense Mutations with Sensitivity to Tyrosine Kinase Inhibitors. EMBO Mol. Med. 2023, 15, e17367. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, X.; Chen, H.; Bao, H.; Wu, X.; Wang, H.; Bao, H.; Pang, J.; Wang, S.; Wang, J. Mechanisms of Resistance to Tyrosine Kinase Inhibitors in ROS1 Fusion-Positive Nonsmall Cell Lung Cancer. Clin. Chem. 2024, 70, 629–641. [Google Scholar] [CrossRef] [PubMed]
- AUGTYROTM (Repotrectinib). Available online: https://accessdata.fda.gov/drugsatfda_docs/label/2023/218213s000lbl.pdf (accessed on 15 March 2024).
- Pizzutilo, E.G.; Agostara, A.G.; Roazzi, L.; Romanò, R.; Motta, V.; Lauricella, C.; Marrapese, G.; Cerea, G.; Signorelli, D.; Veronese, S.M.; et al. Repotrectinib Overcomes F2004V Resistance Mutation in ROS1-Rearranged NSCLC: A Case Report. JTO Clin. Res. Rep. 2023, 4, 100555. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Shaverdashvili, K.; Mino-Kenudson, M.; Digumarthy, S.R.; Do, A.; Liu, A.; Gainor, J.F.; Lennerz, J.K.; Burns, T.F.; Lin, J.J. Lorlatinib and Capmatinib in a ROS1-Rearranged NSCLC with MET-Driven Resistance: Tumor Response and Evolution. NPJ Precis. Oncol. 2023, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.-J.; Lai, Y.-H.; Manne, R.K.; Tsai, Y.-S.; Sarbassov, D.; Lin, H.-K. Akt: A Key Transducer in Cancer. J. Biomed. Sci. 2022, 29, 76. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Dean, E.J.; Bando, H.; El-Khoueiry, A.B.; Pérez-Fidalgo, J.A.; Mita, A.; et al. AKT Inhibition in Solid Tumors with AKT1 Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Calzone, F.J.; Cajulis, E.; Chung, Y.-A.; Tsai, M.-M.; Mitchell, P.; Lu, J.; Chen, C.; Sun, J.; Radinsky, R.; Kendall, R.; et al. Epitope-Specific Mechanisms of IGF1R Inhibition by Ganitumab. PLoS ONE 2013, 8, e55135. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.-L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A Phase 3 Randomized, Double-Blind, Placebo-Controlled Trial of Ganitumab or Placebo in Combination with Gemcitabine as First-Line Therapy for Metastatic Adenocarcinoma of the Pancreas: The GAMMA Trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Krailo, M.D.; Glade-Bender, J.; Buxton, A.; Laack, N.; Randall, R.L.; Chen, H.X.; Seibel, N.L.; Boron, M.; Terezakis, S.; et al. Randomized Phase III Trial of Ganitumab with Interval-Compressed Chemotherapy for Patients with Newly Diagnosed Metastatic Ewing Sarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2023, 41, 2098–2107. [Google Scholar] [CrossRef]
- Choi, M.Y.; Widhopf, G.F.; Wu, C.C.N.; Cui, B.; Lao, F.; Sadarangani, A.; Cavagnaro, J.; Prussak, C.; Carson, D.A.; Jamieson, C.; et al. Pre-Clinical Specificity and Safety of UC-961, a First-in-Class Monoclonal Antibody Targeting ROR1. Clin. Lymphoma Myeloma Leuk. 2015, 15, S167–S169. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Kaufmann, G.F.; Breitmeyer, J.B.; Dickson, K.-A.; Marsh, D.J.; Ford, C.E. The Anti-ROR1 Monoclonal Antibody Zilovertamab Inhibits the Proliferation of Ovarian and Endometrial Cancer Cells. Pharmaceutics 2022, 14, 837. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.-K.; Qin, S.; Yamaguchi, K.; Kim, I.-H.; Saeed, A.; Oh, S.C.; Li, J.; Turk, H.M.; et al. Bemarituzumab in Patients with FGFR2b-Selected Gastric or Gastro-Oesophageal Junction Adenocarcinoma (FIGHT): A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Study. Lancet Oncol. 2022, 23, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Jumapili, N.A.; Zivalj, M.; Barthelmess, R.M.; Raes, G.; De Groof, T.W.M.; Devoogdt, N.; Stijlemans, B.; Vincke, C.; Van Ginderachter, J.A. A Few Good Reasons to Use Nanobodies for Cancer Treatment. Eur. J. Immunol. 2023, 53, 2250024. [Google Scholar] [CrossRef] [PubMed]
- Puttemans, J.; Dekempeneer, Y.; Eersels, J.L.; Hanssens, H.; Debie, P.; Keyaerts, M.; Windhorst, A.D.; Van Der Aa, F.; Lecocq, Q.; Breckpot, K.; et al. Preclinical Targeted α- and Β−-Radionuclide Therapy in HER2-Positive Brain Metastasis Using Camelid Single-Domain Antibodies. Cancers 2020, 12, 1017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xing, Y.; Liu, C.; Ma, S.; Huang, W.; Cheng, Z.; Zhao, J. Detection of HER2 Expression Using 99mTc-NM-02 Nanobody in Patients with Breast Cancer: A Non-Randomized, Non-Blinded Clinical Trial. Breast Cancer Res. 2024, 26, 40. [Google Scholar] [CrossRef] [PubMed]
- D’Huyvetter, M.; Vos, J.D.; Caveliers, V.; Vaneycken, I.; Heemskerk, J.; Duhoux, F.P.; Fontaine, C.; Vanhoeij, M.; Windhorst, A.D.; van der Aa, F.; et al. Phase I Trial of 131I-GMIB-Anti-HER2-VHH1, a New Promising Candidate for HER2-Targeted Radionuclide Therapy in Breast Cancer Patients. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, 62, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luan, L.; Liu, X.; Jiang, D.; Deng, J.; Xu, J.; Yuan, Y.; Xing, J.; Chen, B.; Xing, D.; et al. A Novel Nanobody-Based HER2-Targeting Antibody Exhibits Potent Synergistic Antitumor Efficacy in Trastuzumab-Resistant Cancer Cells. Front. Immunol. 2023, 14, 1292839. [Google Scholar] [CrossRef]
- Khirehgesh, M.R.; Sharifi, J.; Safari, F.; Akbari, B. Immunotoxins and Nanobody-Based Immunotoxins: Review and Update. J. Drug Target. 2021, 29, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Narbona, J.; Hernández-Baraza, L.; Gordo, R.G.; Sanz, L.; Lacadena, J. Nanobody-Based EGFR-Targeting Immunotoxins for Colorectal Cancer Treatment. Biomolecules 2023, 13, 1042. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D.; Bhin, J.; Yemelyanenko, J.; Kas, S.M.; Rolfs, F.; Lutz, C.; Lee, J.K.; Klarenbeek, S.; Silverman, I.M.; Annunziato, S.; et al. Truncated FGFR2 Is a Clinically Actionable Oncogene in Multiple Cancers. Nature 2022, 608, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Montoya, S.; Bourcier, J.; Noviski, M.; Lu, H.; Thompson, M.C.; Chirino, A.; Jahn, J.; Sondhi, A.K.; Gajewski, S.; Tan, Y.S.; et al. Kinase-Impaired BTK Mutations Are Susceptible to Clinical-Stage BTK and IKZF1/3 Degrader NX-2127. Science 2024, 383, eadi5798. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating P53-Based Therapies for Cancer into the Clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef]
- Lu, X.; Jin, J.; Wu, Y.; Liu, X.; Liang, X.; Lin, J.; Sun, Q.; Qin, J.; Zhang, W.; Luan, X. Progress in RAS-Targeted Therapeutic Strategies: From Small Molecule Inhibitors to Proteolysis Targeting Chimeras. Med. Res. Rev. 2024, 44, 812–832. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zhu, J.; Li, J.; Zhan, F.; Yao, H.; Xu, J.; Xu, S. Small-Molecule Hydrophobic Tagging: A Promising Strategy of Druglike Technology for Targeted Protein Degradation: Miniperspective. J. Med. Chem. 2023, 66, 10917–10933. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG System for Immediate and Target-Specific Protein Degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, R.; Xu, F.; Yang, K.; Zheng, L.; Zhao, P.; Shi, G.; Dai, L.; Xu, C.; Yu, L.; et al. Beyond Canonical PROTAC: Biological Targeted Protein Degradation (bioTPD). Biomater. Res. 2023, 27, 72. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Imamura, R.; Suga, H.; Matsumoto, K.; Sakai, K. Cyclic Peptide-Based Biologics Regulating HGF-MET. Int. J. Mol. Sci. 2020, 21, 7977. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, X.; Xie, Y.; Wang, Y. What Influences the Activity of Degrader−Antibody Conjugates (DACs). Eur. J. Med. Chem. 2024, 268, 116216. [Google Scholar] [CrossRef] [PubMed]
- C4 Therapeutics Announces FDA Clearance of Investigational New Drug Application for CFT8919, an Orally Bioavailable BiDACTM Degrader Targeting EGFR L858R for Non-Small Cell Lung Cancer–C4 Therapeutics, Inc. Available online: https://ir.c4therapeutics.com/news-releases/news-release-details/c4-therapeutics-announces-fda-clearance-investigational-new-0/ (accessed on 27 March 2024).
- Park, E.S.; Ahn, J.Y.; Baddour, J.; Chaturvedi, P.; Chiu, M.I.; Cole, K.S.; Crystal, A.S.; Duplessis, M.; Fisher, S.L.; Good, A.C.; et al. Preclinical Evaluation of CFT8919 as a Mutant Selective Degrader of EGFR with L858R Activating Mutations for the Treatment of Non-Small Cell Lung Cancer; C4 Therapeutics Inc.: Watertown, MA, USA, 2021. [Google Scholar]
- Ubix Therapeutics. Available online: http://www.ubixtrx.com/news/press/59 (accessed on 27 March 2024).
- BGB-16673 Delivers Responses with a Tolerable Safety Profile across R/R B-Cell Malignancies. Available online: https://www.onclive.com/view/bgb-16673-delivers-reponses-with-a-tolerable-safety-profile-across-r-r-b-cell-malignancies (accessed on 27 March 2024).
- McKean, M.; Spira, A.I.; Rosen, E.; Subbiah, V.; Moreno, V.; Gambardella, V.; Vieito, M.; Saavedra, O.; Cousin, S.; Cassier, P.A.; et al. A Phase 1/2 Study of CFT1946, a Novel, Bifunctional Degradation Activating Compound (BIDAC) Degrader, of Mutant BRAF V600 as Monotherapy and in Combination with Trametinib, in Mutant BRAF V600 Solid Tumors. J. Clin. Oncol. 2023, 41, TPS3163. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, S.; Han, S.; Zhao, Y.; Yu, C.; Liu, H.; Li, N. Targeted Protein Degrader Development for Cancer: Advances, Challenges, and Opportunities. Trends Pharmacol. Sci. 2023, 44, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Cantrill, C.; Chaturvedi, P.; Rynn, C.; Petrig Schaffland, J.; Walter, I.; Wittwer, M.B. Fundamental Aspects of DMPK Optimization of Targeted Protein Degraders. Drug Discov. Today 2020, 25, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, T.; Cornu, M.; Schwalen, F.; Since, M.; Kieffer, C.; Voisin-Chiret, A.S. Molecular Glue Degraders: Exciting Opportunities for Novel Drug Discovery. Expert. Opin. Drug Discov. 2024, 19, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.; Krönke, J. The Molecular Mechanism of Thalidomide Analogs in Hematologic Malignancies. J. Mol. Med. 2016, 94, 1327–1334. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Liu, M.; Zhang, M.; Jia, X. NEK7: A New Target for the Treatment of Multiple Tumors and Chronic Inflammatory Diseases. Inflammopharmacology 2022, 30, 1179–1187. [Google Scholar] [CrossRef]
- Captor Therapeutics. Available online: https://captortherapeutics.com/pipeline-en (accessed on 28 March 2024).
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.-D.; Manojkumar, M.; Bunker, R.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK Inhibitor CR8 Acts as a Molecular Glue Degrader That Depletes Cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, L.; Ma, B.; Liu, T.; Wang, Z.; Ye, Q.; Peng, Y.; Wang, B.; Chen, Y.; Xu, S.; et al. MYC Induces CDK4/6 Inhibitors Resistance by Promoting pRB1 Degradation. Nat. Commun. 2024, 15, 1871. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-Targeting Chimaeras for Degradation of Extracellular Proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Ahn, G.; Riley, N.M.; Kamber, R.A.; Wisnovsky, S.; Moncayo Von Hase, S.; Bassik, M.C.; Banik, S.M.; Bertozzi, C.R. Elucidating the Cellular Determinants of Targeted Membrane Protein Degradation by Lysosome-Targeting Chimeras. Science 2023, 382, eadf6249. [Google Scholar] [CrossRef] [PubMed]
- Vartak, R.; Deore, B.; Sanhueza, C.A.; Patel, K. Cetuximab-Based PROteolysis Targeting Chimera for Effectual Downregulation of NSCLC with Varied EGFR Mutations. Int. J. Biol. Macromol. 2023, 252, 126413. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, B.; Yang, N.; Xu, T.; Zheng, Z. The next Generation of EGFR Inhibitors: A Patenting Perspective of PROTACs Based EGFR Degraders. Expert Opin. Ther. Pat. 2023, 33, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.H.; Kim, H.Y.; Lee, M.J.; Heo, A.J.; Park, D.Y.; Lim, S.; Shin, S.; Ganipisetti, S.; Yang, W.S.; Jung, C.A.; et al. The AUTOTAC Chemical Biology Platform for Targeted Protein Degradation via the Autophagy-Lysosome System. Nat. Commun. 2022, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhao, X.; Wu, D.; Jia, S.; Liu, C.; Cheng, Z.; Huang, F.; Chen, Y.; Lu, T.; Lu, S. Hydrophobic Tag-Based Protein Degradation: Development, Opportunity and Challenge. Eur. J. Med. Chem. 2023, 260, 115741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; Kumru, K. Extracellular Targeted Protein Degradation: An Emerging Modality for Drug Discovery. Nat. Rev. Drug Discov. 2024, 23, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Kailasam Natesan, V.; Kuppannagounder Pitchaimuthu, E. Structure-Based Drug Design and Molecular Dynamics Studies of an Allosteric Modulator Targeting the Protein–Protein Interaction Site of PDK1. J. Mol. Model. 2024, 30, 51. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, F.; Migliaccio, A. VAL 201—An Inhibitor of Androgen Receptor-Associated Src and a Potential Treatment of Castration-Resistant Prostate Cancer. Eur. Oncol. Haematol. 2012, 8, 32–35. [Google Scholar] [CrossRef]
- Clinical Trials Register-Search for EUCTR2013-004009-25-GB. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=EUCTR2013-004009-25-GB (accessed on 31 March 2024).
- ValiRx Plc A Phase I/II, Dose Escalation Study to Assess the Safety and Tolerability of VAL201 in Patients with Advanced or Metastatic Prostate Cancer and Other Advanced Solid Tumours. 2021. Available online: https://clinicaltrials.gov/ (accessed on 31 March 2024).
- Mallick, M.; Yoithap Prabhunath, T.R.; Kumari, S.; Sobhia, M.E. An in Silico Study of Protein-Protein Interactions and Design of Novel Peptides for TrkA in Ameloblastoma. J. Biomol. Struct. Dyn. 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haines, E.; Catterall, R.; De Jesus, V.; Hidalgo, D.; Cavanaugh, J.; Sanchez-Martin, M.; Manna, J.D.; Burke, M.; Li, B.; Wessel, S.R.; et al. Abstract 3296: IK-595, a Best-in-Class MEK-RAF Molecular Glue, Drives Broad and Potent Anti-Tumor Activity across RAS-MAPK Pathway-Altered Cancers as a Monotherapy and in Combination. Cancer Res. 2024, 84, 3296. [Google Scholar] [CrossRef]
- Quade, B.; Ryan, M.; Swalm, B.; Cohen, S.; Fang, Z.; Ozen, A.; Huang, X.; Dar, A.C.; Han, Y.; Hoeflich, K.P.; et al. Abstract A086: NST-628 Is a Novel Molecular Glue That Inhibits Signaling and Pathway Reactivation in Oncogenic RAS-MAPK Cancers. Mol. Cancer Ther. 2023, 22, A086. [Google Scholar] [CrossRef]
- Gao, G.; Li, J.; Cao, Y.; Li, X.; Qian, Y.; Wang, X.; Li, M.; Qiu, Y.; Wu, T.; Wang, L.; et al. Design, Synthesis, and Biological Evaluation of Novel 4,4′-Bipyridine Derivatives Acting as CDK9-Cyclin T1 Protein-Protein Interaction Inhibitors against Triple-Negative Breast Cancer. Eur. J. Med. Chem. 2023, 261, 115858. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Guo, Y.; Xue, Z.; Guo, Z.; Wang, Z.; Lin, D.; Zhang, H.; Pan, H.; Zhang, X.; Yin, F.; et al. Small-Molecule Inhibitor Targeting the Hsp70-Bim Protein–Protein Interaction in CML Cells Overcomes BCR-ABL-Independent TKI Resistance. Leukemia 2021, 35, 2862–2874. [Google Scholar] [CrossRef]
- Matos, B.; Howl, J.; Jerónimo, C.; Fardilha, M. The Disruption of Protein-Protein Interactions as a Therapeutic Strategy for Prostate Cancer. Pharmacol. Res. 2020, 161, 105145. [Google Scholar] [CrossRef]
- Huang, L.-C.; Taujale, R.; Gravel, N.; Venkat, A.; Yeung, W.; Byrne, D.P.; Eyers, P.A.; Kannan, N. KinOrtho: A Method for Mapping Human Kinase Orthologs across the Tree of Life and Illuminating Understudied Kinases. BMC Bioinf. 2021, 22, 446. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.D.; Knatko, E.; Moore, W.J.; Swedlow, J.R. Mitotic Mechanics: The Auroras Come into View. Curr. Opin. Cell Biol. 2003, 15, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Kumar, M.; Saifi, S.; Rawat, S.; Ethayathulla, A.S.; Kaur, P. A Comprehensive Review on Role of Aurora Kinase Inhibitors (AKIs) in Cancer Therapeutics. Int. J. Biol. Macromol. 2024, 265, 130913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Bi, M.; Su, C.; Fang, Y.; Wang, Z.; Yuan, Y.; Du, X.; Lv, T.; Li, Y. Phase Ib/IIa Study Assessing the Safety and Efficacy of AL8326 Monotherapy in Patients with ≥3rd Line Small Cell Lung Cancer (SCLC) Treatment. J. Clin. Oncol. 2023, 41, 8585. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Xu, B.; Dumbrava, E.E.; Fu, S.; Karp, D.D.; Meric-Bernstam, F.; Hong, D.S.; Rodon, J.A.; Tsimberidou, A.M.; Raghav, K.; et al. First-In-Human Phase I Study of Tinengotinib (TT-00420), a Multiple Kinase Inhibitor, as a Single Agent in Patients with Advanced Solid Tumors. Oncologist 2024, 29, e514–e525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gong, J.; Guo, H.; Han, W.; Cao, B.; Zhang, J.; Niu, Z.; Cheng, Y.; He, C.; Fan, J.; et al. 670P Preliminary Efficacy and Safety of Tinengotinib (TT-00420) Monotherapy in Chinese Patients (Pts) with Advanced Solid Tumors: Results from a Phase Ib/II Study. Ann. Oncol. 2023, 34, S471. [Google Scholar] [CrossRef]
- Alli, V.J.; Yadav, P.; Suresh, V.; Jadav, S.S. Synthetic and Medicinal Chemistry Approaches Toward WEE1 Kinase Inhibitors and Its Degraders. ACS Omega 2023, 8, 20196–20233. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Hendrickson, A.E.W.; et al. Adavosertib plus Gemcitabine for Platinum-Resistant or Platinum-Refractory Recurrent Ovarian Cancer: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Lee, N.K.; Saad, S.E.; Fong, I.L. Clinical Translation for Targeting DNA Damage Repair in Non-Small Cell Lung Cancer: A Review. Transl. Lung Cancer Res. 2024, 13, 375–397. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a Cell Cycle Checkpoint Kinase 1 and 2 Inhibitor, in BRCA Wild-Type Recurrent High-Grade Serous Ovarian Cancer: A First-in-Class Proof-of-Concept Phase 2 Study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.H.; Chu, Q.S.; Bouganim, N.; Jonker, D.J.; Batist, G.; Buhlaiga, N.J.; Lheureux, S.; Spratlin, J.L.; Alcindor, T.; Smith, P.S.; et al. A Phase Ib Study of Oral Chk1 Inhibitor LY2880070 as Monotherapy in Patients with Advanced or Metastatic Cancer. J. Clin. Oncol. 2020, 38, 3579. [Google Scholar] [CrossRef]
- Xue, C.; Yao, Q.; Gu, X.; Shi, Q.; Yuan, X.; Chu, Q.; Bao, Z.; Lu, J.; Li, L. Evolving Cognition of the JAK-STAT Signaling Pathway: Autoimmune Disorders and Cancer. Signal Transduct. Target. Ther. 2023, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Li, S.; Ma, J.; Dai, X.; Lu, J. Deciphering JAK/STAT Signaling Pathway: A Multifaceted Approach to Tumorigenesis, Progression and Therapeutic Interventions. Int. Immunopharmacol. 2024, 131, 111846. [Google Scholar] [CrossRef]
- Munster, P.; Iannotti, N.; Cho, D.C.; Kirkwood, J.M.; Villaruz, L.C.; Gibney, G.T.; Hodi, F.S.; Mettu, N.B.; Jones, M.; Bowman, J.; et al. Combination of Itacitinib or Parsaclisib with Pembrolizumab in Patients with Advanced Solid Tumors: A Phase I Study. Cancer Res. Commun. 2023, 3, 2572–2584. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.; Palandri, F.; Baer, M.R.; Quick, D.; Kiladjian, J.J.; Martinelli, G.; Verma, A.; Hamid, O.; Walgren, R.; Pitou, C.; et al. Phase 2 Study of Gandotinib (LY2784544) in Patients with Myeloproliferative Neoplasms. Leuk. Res. 2018, 71, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Faya Castillo, J.E.; Zapata Dongo, R.J.; Wong Chero, P.A.; Infante Varillas, S.F. Mitoxantrone and Abacavir: An ALK Protein-Targeted in Silico Proposal for the Treatment of Non-Small Cell Lung Cancer. PLoS ONE 2024, 19, e0295966. [Google Scholar] [CrossRef] [PubMed]
- Muthuraj, R.; Gopal, D.; Ahmed, I.; Chandrasekaran, J. Insightful T-SNE Guided Exploration Spotlighting Palbociclib and Ribociclib Analogues as Novel WEE1 Kinase Inhibitory Candidates. J. Biomol. Struct. Dyn. 2024, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, P.; Siddiqui, N.F.; Thakur, S.; Jadhav, H. FDA Approved Fused-Pyrimidines as Potential PI3K Inhibitors: A Computational Repurposing Approach. J. Biomol. Struct. Dyn. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- von Itzstein, M.S.; Gerber, D.E. Resistance Mechanisms in ROS1-Positive Lung Cancer: New Insight into a Rare but Clinically Important Entity. Clin. Chem. 2024, 70, 571–573. [Google Scholar] [CrossRef]
- Shaikh, M.; Shinde, Y.; Pawara, R.; Noolvi, M.; Surana, S.; Ahmad, I.; Patel, H. Emerging Approaches to Overcome Acquired Drug Resistance Obstacles to Osimertinib in Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 1008–1046. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Lu, C.; Hu, C.; Dou, Y.; Kang, J.; Lin, C.; Wu, D.; Jiang, W.; Yin, G.; He, Y. Brigatinib, a Newly Discovered AXL Inhibitor, Suppresses AXL-Mediated Acquired Resistance to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer. Acta Pharmacol. Sin. 2024. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Concannon, K.; Li, J.; Zhang, J.; Heymach, J.V.; Le, X. Emerging Therapeutics and Evolving Assessment Criteria for Intracranial Metastases in Patients with Oncogene-Driven Non-Small-Cell Lung Cancer. Nat. Rev. Clin. Oncol. 2023, 20, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Badhan, R.; Chenel, M.; Penny, J. Development of a Physiologically-Based Pharmacokinetic Model of the Rat Central Nervous System. Pharmaceutics 2014, 6, 97–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; de Braud, F.G.; Maur, M.; Loong, H.H.; Shaw, A.T.; Vansteenkiste, J.F.; John, T.; Liu, G.; Lolkema, M.P.; Selvaggi, G.; et al. Ceritinib plus Nivolumab in Patients with Advanced ALK-Rearranged Non–Small Cell Lung Cancer: Results of an Open-Label, Multicenter, Phase 1B Study. J. Thorac. Oncol. 2020, 15, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Starzer, A.M.; Wolff, L.; Popov, P.; Kiesewetter, B.; Preusser, M.; Berghoff, A.S. The More the Merrier? Evidence and Efficacy of Immune Checkpoint- and Tyrosine Kinase Inhibitor Combinations in Advanced Solid Cancers. Cancer Treat. Rev. 2024, 125, 102718. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.-C. Targeting Tumour Microenvironment by Tyrosine Kinase Inhibitor. Mol. Cancer 2018, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Lim, K.-H.; McWilliams, R.; Suresh, R.; Lockhart, A.C.; Brown, A.; Breden, M.; Belle, J.I.; Herndon, J.; Bogner, S.J.; et al. Defactinib, Pembrolizumab, and Gemcitabine in Patients with Advanced Treatment Refractory Pancreatic Cancer: A Phase I Dose Escalation and Expansion Study. Clin. Cancer Res. 2022, 28, 5254–5262. [Google Scholar] [CrossRef] [PubMed]


| Targets | Compound Name | Alternatives | Drug Type | Indications in Research | Highest Study Phase |
|---|---|---|---|---|---|
| EGFR | Futuximab/Modotuximab | DS-992/DS-1024 | Monoclonal antibody | Solid tumor, metastatic colorectal cancer | Phase 3 Company pipeline Symphogen A/S (Lyngby, Denmark) |
| HER2 | HLX-22 | AC-101, HLX-22 | Monoclonal antibody | Gastric cancer, breast cancer, metastatic gastric cancer | Phase 3 Company pipeline AbClon, Inc. (Seoul, Republic of Korea). |
| EGFR | Pimurutamab | Monoclonal antibody | Cutaneous squamous cell carcinoma, squamous NSCLC | Phase 3 Company pipeline Shanghai Henlius Biotech, Inc. (Shanghai, China). | |
| ROR1 | Zilovertamab | Cirmtuzumab, UC-961, ZILO-301 | Monoclonal antiboby | Mantle cell lymphoma, chronic lymphocytic leukemia | Phase 3 NCT05431179 University of California, (Los Angeles, CA, USA) |
| VEGFR2 | Gentuximab | GenSci-043 GenSci043 GenSci043-01 | Monoclonal antiboby | Advanced gastroesophageal junction adenocarcinoma, EGFR T790M mutation, NSCLC | Phase 3 CTR20220815 Changchun Genescience Pharmaceuticals Co., Ltd. (Changchun, China). |
| FGFR2 | Bemarituzumab | FPA-144 | Monoclonal antiboby | FGFR2b-positive gastroesophageal junction cancer | Phase 3 CTR20233946 Five Prime Therapeutics, Inc. (San Francisco, CA, USA). |
| IGF-1R | Ganitumab | AMG-479 | Monoclonal antiboby | Bone metastases, Ewing’s sarcoma | Phase 3 NCT02306161 Amgen, Inc. (Thousand Oaks, CA, USA) |
| EGFR | JMT-101 | JMT101 | Monoclonal antiboby | Solid tumors, squamous non-small-cell lung cancer | Phase 2/3 NCT06319313 Shanghai JMT Biological Technology Co., Ltd. (Shanghai, China). |
| PDGFRβ | 68Ga-BOT1712 | Nanobody | Colon cancer, liver cirrhosis | Preclinical Company pipeline Cortalix BV (Groningen, The Netherlands) | |
| HER2 | RAD202 | Nanobody | HER2-positive breast cancer | Preclinical Company pipeline Radiopharm Theranostics, Ltd. (Carlton, VIC, Australia). | |
| PDGFRβ | 177Lu-BOT1712 | Nanobody | Colon cancer | Preclinical Company pipeline Cortalix BV |
| Target | Compound Name | Drug Type | Indications in Research | Highest Clinical Phase |
|---|---|---|---|---|
| NTRK | CG-001419 | PROTACs | Solid tumors | Phase 1/2 Cullgen, Inc. (San Diego, CA, USA). CXHL2200331 * |
| EGFR | HSK40118 | PROTACs | EGFR-positive NSCLC | Phase 1 Haisco Pharmaceutical Group Co., Ltd. (Chengdu, China). CTR20230926 |
| GSPT1 × HER2 | ORM-5029 | PROTACs | HER2-positive solid tumors or breast cancer | Phase 1 Orum Therapeutics, Inc. (Daejeon, Republic of Korea). NCT05511844 |
| EGFR L858R | CFT-8919 | PROTACs | EGFR-positive NSCLC | Investigational new drug (IND) by FDA C4 Therapeutics, Inc. (Watertown, MA, USA). Company pipeline |
| BTK | BGB-16673 | PROTACs | Chronic lymphocytic leukemia, B-cell lymphoma | Phase 1 BeiGene Ltd. (Cambridge, MA, USA). NCT05006716 |
| BRAF, BTK | CFT-1946 | PROTACs | BRAF V600 mutation-positive tumor, brain metastases, colorectal cancer | Phase 2 C4 Therapeutics, Inc. (Watertown, MA, USA). NCT05668585 |
| BTK, BTK C481S | AC-0676 | PROTACs | B-cell malignancies, tumors, chronic lymphocytic leukemia | Phase 1 Accutar Biotechnology, Inc. (Shanghai, China). NCT05780034 |
| BTK | HSK-29116 | PROTACs | B-cell lymphoma | Phase 1 Sichuan Haisco Pharmaceutical Co., Ltd. (Chengdu, China). NCT04861779 |
| EGFR | HSK40118 | PROTACs | EGFR-positive NSCLC | Phase 1 Haisco Pharmaceutical Group Co., Ltd. (Chengdu, China). CTR20230926 |
| BTK | HZ-Q1070 | PROTACs | B-cell malignancies | Phase 1 Hangzhou Hertz Pharmaceutical Co., Ltd. (Hangzhou, China). CTR20240471 |
| BTK | UBX-303-1 | PROTACs | Chronic lymphocytic leukemia | Clinical Application for approval Ubix Therapeutics Co., Ltd. (Seoul, Republic of Korea). Company pipeline |
| MEK, Raf | IK-595 | Molecular glue | Colorectal cancer | Phase 1 Ikena Oncology, Inc. (Boston, MA, USA). NCT06270082 |
| MAPK, Ras | NST-628 | Molecular glue | Glioma, melanoma | Phase 1 Nested Therapeutics, Inc. (Cambridge, MA, USA). NCT06326411 |
| CDK4, CDK6, c- Myc | A80.2 HCI | Molecular glue | Bladder cancer, breast cancer | Preclinical Suzhou Kintor Pharmaceuticals, Inc. (Suzhou, China). Company pipeline |
| c-Met | HIP-8 | Polypeptide | Tumors | Preclinical Kanazawa University (Ishikawa, Japan) [147] |
| Compound Name | Target | Mechanism of Action | Indications in Research | Clincial Phase | Clinical Trail |
|---|---|---|---|---|---|
| AL-8326 | Aurora B, FGFRs, VEGFR | Aurora B inhibitors, FGFR antagonists, VEGFR antagonists | Small-cell lung cancer bile duct tumor, bladder cancer | Phase 3 | CTR20233349 |
| Chiauranib | Aurora B, CSF-1R | Aurora B inhibitor, CSF-1R antagonist, VEGFR junction inhibitor | Relapsing drug-resistant ovarian cancer, soft tissue sarcoma | Phase 3 | Chiauranib |
| Tinengotinib | Aurora A, Aurora B, CSF-1R | Aurora A inhibitors, Aurora B inhibitors, CSF-1R antagonists | Bile duct epithelial carcinoma, triple-negative breast cancer | Phase 3 | NCT05948475 |
| CEP-11981 | Tie-2, VEGF | Tie-2 antagonists | Adenosquamous carcinoma, gastrointestinal pancreatic neuroendocrine tumor | Phase 2 | NCT05988918 |
| Gandotinib | JAK2 | JAK2 inhibitors | Blood tumor | Phase 2 | NCT01594723 |
| Itacitinib | JAK1 | JAK1 inhibitor | Diffuse large B-cell lymphoma | Phase 2 | NCT03139604 |
| LY-2880070 | CHK1 | CHK1 inhibitors | Ewing sarcoma tumor | Phase 2 | NCT05275426 |
| Onvansertib | PLK1 | PLK1 inhibitors | Small-cell lung cancer, recurrent metastatic colorectal cancer | Phase 2 | NCT05450965 |
| Prexasertib | CHK1 x CHK2 | CHK1 inhibitors, Chk2 inhibitors | Metastatic triple-negative breast cancer | Phase 2 | NCT05548296 |
| Golidocitinib | JAK1 | JAK1 | Peripheral T-cell lymphoma, recurrent T-cell lymphoma | Phase 2 | NCT04105010 |
| Adavosertib | WEE1 | WEE1 inhibitor | Local late-stage clear cell/renal cell carcinoma, metastatic renal cell carcinoma | Phase 2 | NCT03284385 |
| Azenosertib | WEE1 | WEE1 inhibitor | Pancreatic ductal adenocarcinoma, acute myeloid leukemia | Phase 2 | NCT06015659 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Gong, C.; Zhou, H.; Liu, J.; Xia, X.; Ha, W.; Jiang, Y.; Liu, Q.; Xiong, H. Kinase Inhibitors and Kinase-Targeted Cancer Therapies: Recent Advances and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 5489. https://doi.org/10.3390/ijms25105489
Li J, Gong C, Zhou H, Liu J, Xia X, Ha W, Jiang Y, Liu Q, Xiong H. Kinase Inhibitors and Kinase-Targeted Cancer Therapies: Recent Advances and Future Perspectives. International Journal of Molecular Sciences. 2024; 25(10):5489. https://doi.org/10.3390/ijms25105489
Chicago/Turabian StyleLi, Jiahao, Chen Gong, Haiting Zhou, Junxia Liu, Xiaohui Xia, Wentao Ha, Yizhi Jiang, Qingxu Liu, and Huihua Xiong. 2024. "Kinase Inhibitors and Kinase-Targeted Cancer Therapies: Recent Advances and Future Perspectives" International Journal of Molecular Sciences 25, no. 10: 5489. https://doi.org/10.3390/ijms25105489