
Characterization of the Mitogenome of the Genus Dendrocerus Ratzeburg (Hymenoptera: Megaspilidae) with the Specific Designed Primers
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and DNA Extraction
2.2. Sequencing and Assembly
2.3. Mitochondrial Genome Annotation
2.4. Mitochondrial Genome Comparative Analysis
2.5. Phylogenetic Analysis
2.6. Specific Primer Design
3. Results
3.1. Mitogenome Structure and Organization
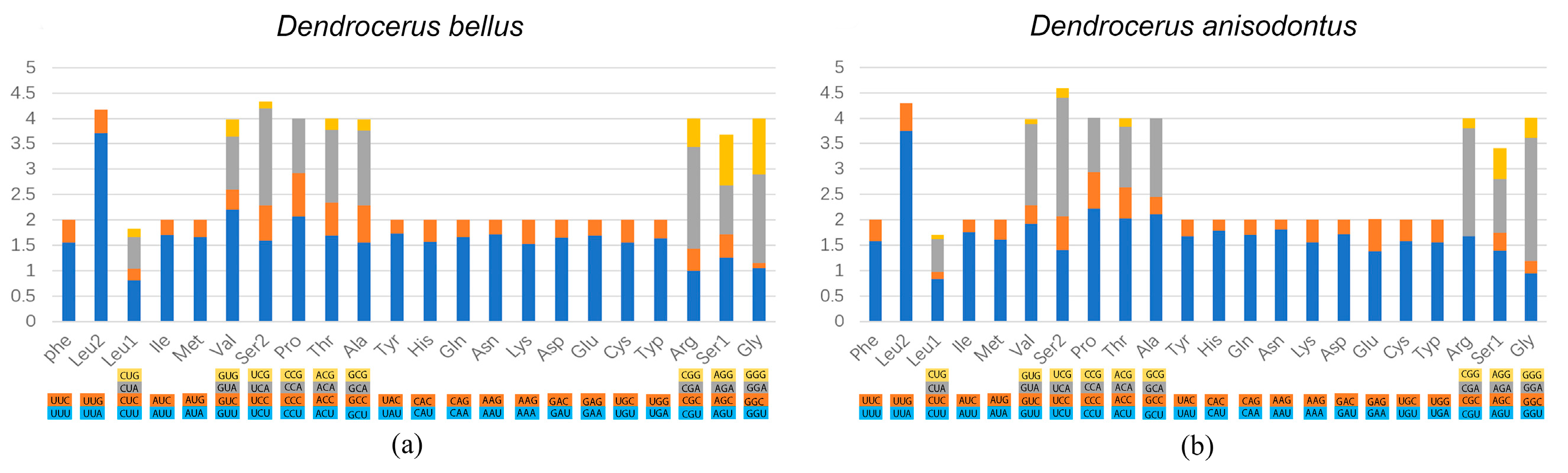
3.2. Protein-Coding Genes
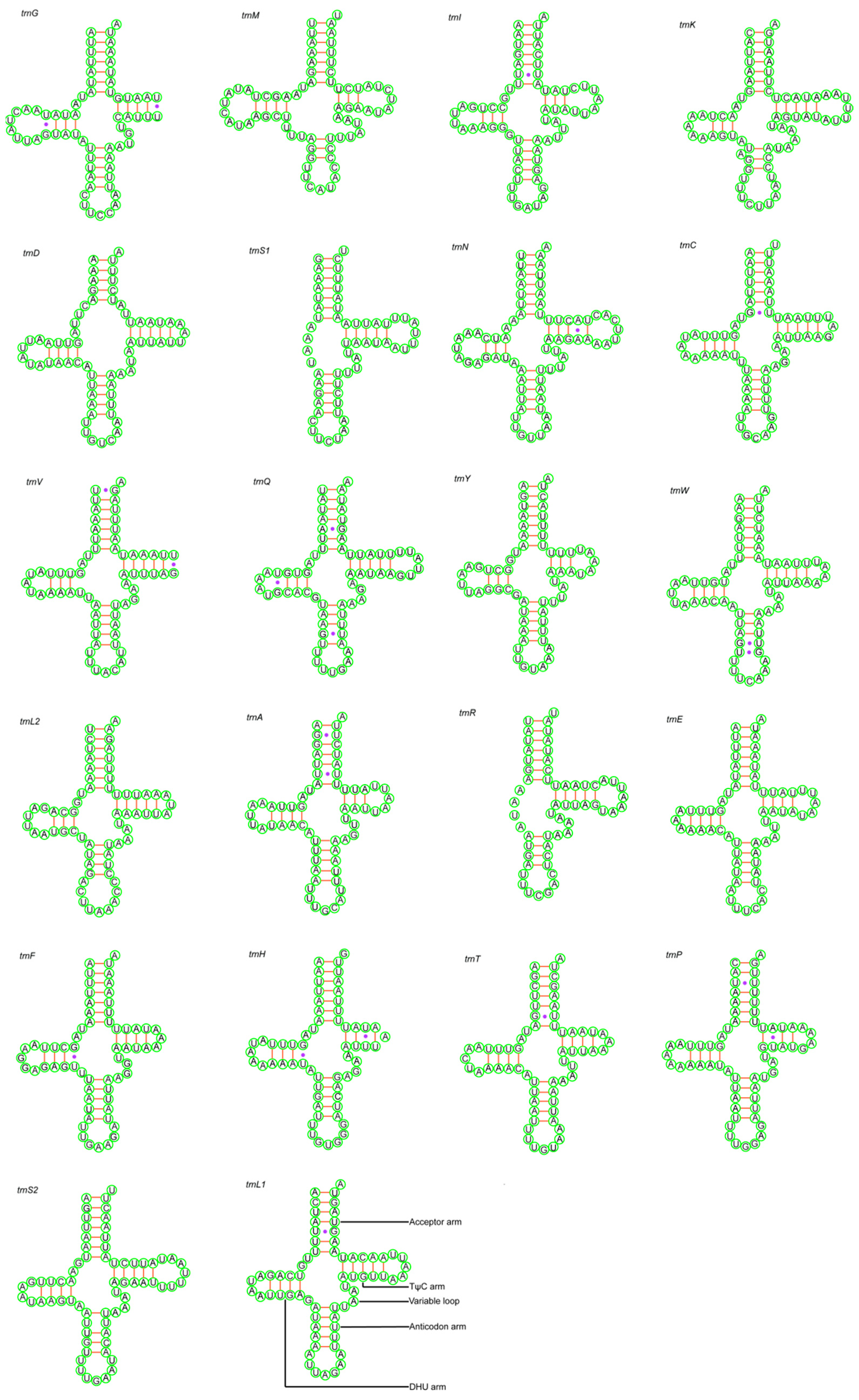
3.3. Transfer RNA and Ribosomal RNA Genes
3.4. Overlap and Gap
3.5. Evolutionary Rate Analysis
3.6. Gene Arrangement
3.7. Phylogenetic Analysis
3.8. Specific Primer Design
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iemma, L.G.R.; Tavares, M.T.; Sousa-Silva, C.R. First record of Dendrocerus carpenteri (Hymenoptera: Megaspilidae) on Aphidius ervi in alfalfa fields in Brazil. Braz. J. Biol. 2016, 76, 1066–1067. [Google Scholar] [CrossRef] [PubMed]
- Dessart, P. Familia Megaspilidae. In Hymenoptera de la Región Neotropical; Hanson, P.E., Gould, I.D., Eds.; Memoirs of the American Entomological Institute: Gainesville, FL, USA, 2006; Volume 77, pp. 232–236. [Google Scholar]
- Blaimer, B.B.; Santos, B.F.; Cruaud, A.; Gates, M.W.; Kula, R.R.; Mikó, I.; Rasplus, J.-Y.; Smith, D.R.; Talamas, E.J.; Brady, S.G.; et al. Key innovations and the diversification of Hymenoptera. Nat. Commun. 2023, 14, 1212. [Google Scholar] [CrossRef] [PubMed]
- Heraty, J.; Ronquist, F.; Carpenter, J.M.; Hawks, D.; Schulmeister, S.; Dowling, A.P.; Murray, D.; Munro, J.; Wheeler, W.C.; Schiff, N.; et al. Evolution of the hymenopteran megaradiation. Mol. Phylogenetics Evol. 2011, 60, 73–88. [Google Scholar] [CrossRef]
- Rasnttsyn, A.P.; Haichun, Z. Early Evolution of Apocrita (Insecta, Hymenoptera) as Indicated by New Findings in the Middle Jurassic of Daohugou, Northeast China. Acta Geol. Sin. -Engl. Ed. 2010, 84, 834–873. [Google Scholar] [CrossRef]
- Tang, P.; Zhu, J.-C.; Zheng, B.-Y.; Wei, S.-J.; Sharkey, M.; Chen, X.-X.; Vogler, A.P. Mitochondrial phylogenomics of the Hymenoptera. Mol. Phylogenetics Evol. 2019, 131, 8–18. [Google Scholar] [CrossRef]
- Klopfstein, S.; Vilhelmsen, L.; Heraty, J.M.; Sharkey, M.; Ronquist, F. The hymenopteran tree of life: Evidence from protein-coding genes and objectively aligned ribosomal data. PLoS ONE 2013, 8, e69344. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Gibson, T.; Dowton, M. Evolutionary dynamics of the mitochondrial genome in the evaniomorpha (hymenoptera)—A group with an intermediate rate of gene rearrangement. Genome Biol. Evol. 2014, 6, 1862–1874. [Google Scholar] [CrossRef]
- Peters, R.S.; Krogmann, L.; Mayer, C.; Donath, A.; Gunkel, S.; Meusemann, K.; Kozlov, A.; Podsiadlowski, L.; Petersen, M.; Lanfear, R.; et al. Evolutionary History of the Hymenoptera. Curr. Biol. 2017, 27, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, M.J.; Carpenter, J.M.; Vilhelmsen, L.; Heraty, J.; Liljeblad, J.; Dowling, A.P.; Schulmeister, S.; Murray, D.; Deans, A.R.; Ronquist, F.; et al. Phylogenetic relationships among superfamilies of Hymenoptera. Cladistics 2012, 28, 80–112. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Èntomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Huang, L.; Sun, H.-Q.; Li, C.-J.; Zhao, W.-X.; Yao, Y.-X. The complete mitochondrial genome of Platygaster robiniae (Hymenoptera: Platygastridae): A novel tRNA secondary structure, gene rearrangements and phylogenetic implications. Int. J. Parasitol. Parasites Wildl. 2022, 18, 249–259. [Google Scholar] [CrossRef]
- Huang, L.-S.; Liu, Z.; Chen, H.-Y.; Chen, X.-X. Two mitochondrial genomes of genus Megacampsomeris Betrem, 1928 (Hymenoptera: Scoliidae) with their phylogenetic implications. J. Asia-Pac. Entomol. 2023, 26, 102067. [Google Scholar] [CrossRef]
- Jasso-Martínez, J.M.; Quicke, D.L.J.; Belokobylskij, S.A.; Santos, B.F.; Fernández-Triana, J.L.; Kula, R.R.; Zaldívar-Riverón, A. Mitochondrial phylogenomics and mitogenome organization in the parasitoid wasp family Braconidae (Hymenoptera: Ichneumonoidea). BMC Ecol. Evol. 2022, 22, 46. [Google Scholar] [CrossRef]
- Mikó, I.; Masner, L.; Johannes, E.; Yoder, M.J.; Deans, A.R. Male terminalia of Ceraphronoidea: Morphological diversity in an otherwise monotonous taxon. Insect Syst. Evol. 2013, 44, 261–347. [Google Scholar] [CrossRef]
- Vasilita, C.; Moser, M.; Krogmann, L. Mission possible: An optimised protocol for the unbarcodable Ceraphronoidea (Hymenoptera). Biodivers. Data J. 2022, 10, e84860. [Google Scholar] [CrossRef]
- Fergusson, N.D.M. A revision of the British species of Dendrocerus Ratzeburg (Hymenoptera: Ceraphronoidea) with a review of their biology as aphid hyperparasites. Bull. Br. Mus. (Natural Hist.) 1980, 41, 255–314. [Google Scholar] [CrossRef]
- Miko, I.; Deans, A. Masner, a new genus of Ceraphronidae (Hymenoptera, Ceraphronoidea) described using controlled vocabularies. ZooKeys 2009, 20, 127–153. [Google Scholar] [CrossRef]
- Wang, X.; Chen, H.; Mikó, I.; Huang, Y.; Zhu, C. Notes on the genus Dendrocerus Ratzeburg (Hymenoptera, Megaspilidae) from China, with description of two new species. J. Hymenopt. Res. 2021, 86, 123–143. [Google Scholar] [CrossRef]
- Shahjahan, R.M.; Hughes, K.J.; Leopold, R.A.; DeVault, J.D. Lower incubation temperature increases yield of insect genomic DNA isolated by the CTAB method. Biotechniques 1995, 19, 332–334. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 February 2020).
- Huang, Y.-X.; Ren, F.-J.; Bartlett, C.R.; Wei, Y.-S.; Qin, D.-Z. Contribution to the mitogenome diversity in Delphacinae: Phylogenetic and ecological implications. Genomics 2020, 112, 1363–1370. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Ma, C.; Yang, P.; Jiang, F.; Chapuis, M.-P.; Shali, Y.; Sword, G.A.; Kang, L. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 2012, 21, 4344–4358. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Foster, P.G.; Jermiin, L.S.; Hickey, D.A. Nucleotide composition bias affects amino acid content in proteins coded by animal mitochondria. J. Mol. Evol. 1997, 44, 282–288. [Google Scholar] [CrossRef]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef]
- Galtier, N.; Roux, C.; Rousselle, M.; Romiguier, J.; Figuet, E.; Glémin, S.; Bierne, N.; Duret, L. Codon Usage Bias in Animals: Disentangling the Effects of Natural Selection, Effective Population Size, and GC-Biased Gene Conversion. Mol. Biol. Evol. 2018, 35, 1092–1103. [Google Scholar] [CrossRef]
- Mao, M.; Austin, A.D.; Johnson, N.F.; Dowton, M. Coexistence of minicircular and a highly rearranged mtDNA molecule suggests that recombination shapes mitochondrial genome organization. Mol. Biol. Evol. 2014, 31, 636–644. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regions | Species | Size (bp) | T% | C% | A% | G% | AT (%) | GC (%) | AT Skew | GC Skew |
|---|---|---|---|---|---|---|---|---|---|---|
| Full genome | Dendrocerus bellus | 15,445 | 40.7 | 12.2 | 40.5 | 6.7 | 81.2 | 18.8 | −0.0025 | −0.2910 |
| Dendrocerus anisodontus | 15,373 | 40.2 | 11.7 | 42.2 | 5.9 | 82.4 | 17.6 | 0.0243 | −0.3295 | |
| PCGs | Dendrocerus bellus | 11,173 | 39.5 | 13.3 | 39.7 | 7.6 | 79.2 | 20.8 | 0.0025 | −0.2727 |
| Dendrocerus anisodontus | 11,214 | 39.5 | 12.6 | 41.4 | 6.6 | 80.9 | 19.1 | 0.0235 | −0.3125 | |
| tRNAs | Dendrocerus bellus | 1349 | 43.5 | 9.3 | 41.6 | 5.6 | 85.1 | 14.9 | −0.0223 | −0.2483 |
| Dendrocerus anisodontus | 1415 | 43.1 | 9.2 | 42.3 | 5.4 | 85.4 | 14.6 | −0.0094 | −0.2603 | |
| rRNAs | Dendrocerus bellus | 2121 | 43.8 | 9.1 | 43.1 | 3.9 | 86.9 | 13.1 | −0.0081 | −0.4000 |
| Dendrocerus anisodontus | 2169 | 41.7 | 9.4 | 45.1 | 3.8 | 86.8 | 13.2 | 0.0392 | −0.4242 |
| Position | Size (bp) | Intergenic Nucleotide | Codon | Strand | |||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| Dendrocerus bellus/Dendrocerus anisodontus | |||||||
| trnG | −/173 | −/237 | −/65 | /+ | |||
| trnM | 264/253 | 328/320 | 65/68 | −/15 | +/+ | ||
| trnI | 326/318 | 389/382 | 64/65 | −3/−3 | +/+ | ||
| COX2 | 410/402 | 1114/1103 | 705/702 | 20/19 | ATT/ATT | TAA/TAA | +/+ |
| trnK | 1116/1132 | 1184/1200 | 69/69 | 1/28 | +/+ | ||
| trnD | 1290/1200 | 1353/1265 | 64/66 | 105/−1 | +/+ | ||
| ATP8 | 1407/1266 | 1568/1424 | 162/159 | 53/− | ATC/ATA | TAA/TAA | +/+ |
| ATP6 | 1565/1425 | 2242/2096 | 678/672 | −4/− | ATA/ATG | TAA/TAA | +/+ |
| COX3 | 2246/2100 | 3043/2897 | 798/798 | 3/3 | ATG/ATG | TAA/TAA | +/+ |
| trnS1 | 3047/2907 | 3101/2963 | 55/57 | 3/9 | +/+ | ||
| trnN | 3102/2964 | 3168/3033 | 67/70 | −/− | +/+ | ||
| trnC | 3166/3031 | 3225/3095 | 60/65 | −3/−3 | −/− | ||
| trnV | 3223/3108 | 3287/3171 | 65/64 | −3/11 | −/− | ||
| trnQ | 3292/3173 | 3359/3239 | 68/67 | 4/1 | −/− | ||
| trnY | 3365/3256 | 3426/3317 | 62/62 | 5/16 | −/− | ||
| trnW | 3436/3330 | 3594/3395 | 69/66 | 9/12 | −/− | ||
| COX1 | 3505/3396 | 5052/4938 | 1548/1543 | −/− | ATG/ATG | TAA/T | +/+ |
| trnL2 | 5055/4939 | 5119/5002 | 65/64 | 2/− | +/+ | ||
| NAD3 | 5147/5032 | 5509/5394 | 363/363 | 27/29 | ATT/ATT | TAA/TAA | +/+ |
| trnA | 5517/5398 | 5582/5460 | 66/63 | 7/3 | +/+ | ||
| trnR | 5582/5463 | 5633/5518 | 52/56 | −1/2 | +/+ | ||
| trnE | 5655/5522 | 5719/5584 | 65/63 | 21/3 | +/+ | ||
| trnF | 5719/5619 | 5781/5681 | 63/63 | −1/34 | −/− | ||
| NAD5 | 5782/5682 | 7453/7350 | 1672/1669 | −/− | ATT/ATT | T/T | −/− |
| trnH | 7455/7351 | 7518/7411 | 64/61 | 1/− | −/− | ||
| NAD4 | 7531/7412 | 8868/8749 | 1338/1338 | 12/− | ATG/ATG | TAA/TAG | −/− |
| NAD4L | 8862/8743 | 9143/9024 | 282/282 | −7/−7 | ATG/ATA | TAA/TAA | −/− |
| trnT | 9152/9029 | 9217/9091 | 66/63 | 8/4 | +/+ | ||
| trnP | 9244/9093 | 9307/9156 | 64/64 | 26/1 | −/− | ||
| NAD6 | 9317/9157 | 9844/9706 | 528/550 | 9/− | ATC/ATA | TAA/T | +/+ |
| CYTB | 9860/9707 | 10,978/10,840 | 1119/1134 | 15/− | ATG/ATG | TAG/TAA | +/+ |
| trnS2 | 10,986/10,847 | 11,053/10,914 | 68/68 | 7/6 | +/+ | ||
| NAD1 | 11,059/10,913 | 12,015/11,872 | 957/960 | 5/−2 | ATA/ATA | TAG/TAA | −/− |
| trnL1 | 12,016/11,873 | 12,083/11,938 | 68/66 | −/− | −/− | ||
| rrnL | 12,084/11,939 | 13,419/13,322 | 1336/1384 | −/− | −/− | ||
| NAD2 | 13,420/13,323 | 14,442/14,366 | 1023/1044 | −/− | ATT/ATA | TAA/TAA | +/+ |
| rrnS | 14,443/14,367 | 15,227/15,151 | 785/785 | −/− | −/+ | ||
| Sequencing Sample Number | Species | Sex | Location | GenBank Accession Number |
|---|---|---|---|---|
| A001 | D. sp.1 | female | Chongqing, Yintiaoling National Nature Reserve | OR578603 |
| A002 | D. sp.1 | female | Chongqing, Yintiaoling National Nature Reserve | OR578604 |
| A003 | D. sp.1 | male | Chongqing, Yintiaoling National Nature Reserve | OR578605 |
| A004 | D. sp.1 | female | Chongqing, Yintiaoling National Nature Reserve | OR578606 |
| A005 | D. sp.1 | female | Chongqing, Yintiaoling National Nature Reserve | OR578607 |
| A006 | D. sp.1 | male | Chongqing, Yintiaoling National Nature Reserve | OR578608 |
| A007 | D. sp.2 | male | Chongqing, Yintiaoling National Nature Reserve | OR578609 |
| A008 | D. sp.3 | female | Chongqing, Yintiaoling National Nature Reserve | OR578610 |
| A009 | D. sp.4 | male | Chongqing, Yintiaoling National Nature Reserve | OR578611 |
| A001 | A002 | A003 | A004 | A005 | A006 | A007 | A008 | A009 | |
|---|---|---|---|---|---|---|---|---|---|
| A001 | |||||||||
| A002 | 0.0000 | ||||||||
| A003 | 0.0015 | 0.0015 | |||||||
| A004 | 0.0124 | 0.0124 | 0.0139 | ||||||
| A005 | 0.0000 | 0.0000 | 0.0015 | 0.0124 | |||||
| A006 | 0.0000 | 0.0000 | 0.0015 | 0.0124 | 0.0000 | ||||
| A007 | 0.1731 | 0.1731 | 0.1712 | 0.1849 | 0.1731 | 0.1731 | |||
| A008 | 0.1529 | 0.1529 | 0.1548 | 0.1546 | 0.1529 | 0.1529 | 0.1937 | ||
| A009 | 0.1752 | 0.1752 | 0.1772 | 0.1811 | 0.1752 | 0.1752 | 0.1715 | 0.2037 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhao, W.; Cui, S.; Su, B.; Huang, Y.; Chen, H. Characterization of the Mitogenome of the Genus Dendrocerus Ratzeburg (Hymenoptera: Megaspilidae) with the Specific Designed Primers. Animals 2024, 14, 1454. https://doi.org/10.3390/ani14101454
Wang X, Zhao W, Cui S, Su B, Huang Y, Chen H. Characterization of the Mitogenome of the Genus Dendrocerus Ratzeburg (Hymenoptera: Megaspilidae) with the Specific Designed Primers. Animals. 2024; 14(10):1454. https://doi.org/10.3390/ani14101454
Chicago/Turabian StyleWang, Xu, Wenjing Zhao, Shanshan Cui, Baoshan Su, Yixin Huang, and Huayan Chen. 2024. "Characterization of the Mitogenome of the Genus Dendrocerus Ratzeburg (Hymenoptera: Megaspilidae) with the Specific Designed Primers" Animals 14, no. 10: 1454. https://doi.org/10.3390/ani14101454