
Hypermethylation of the Gene Body in SRCIN1 Is Involved in Breast Cancer Cell Proliferation and Is a Potential Blood-Based Biomarker for Early Detection and a Poor Prognosis
 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Breast Tissue and Plasma Collection
2.2. Genomic DNA, Circulating Cell-Free DNA and RNA Extraction
2.3. DNA Methylation Array Assay
2.4. Gene-Specific DNA Methylation Assay Using Quantitative Methylation-Specific PCR (qMSP)
2.5. Gene Expression Assays Using Reverse Transcriptase qPCR
2.6. Cell Culture, Counting and RNAi Transfection
2.7. Cell Viability Assay
2.8. RNA Sequencing and Signaling Pathway Analysis
2.9. The Cancer Genome Atlas Portal and Data Analysis
2.10. Statistical Analysis
3. Results
3.1. SRCIN1 Was Identified through Genome-Wide Methylation and RNA Expression Analysis
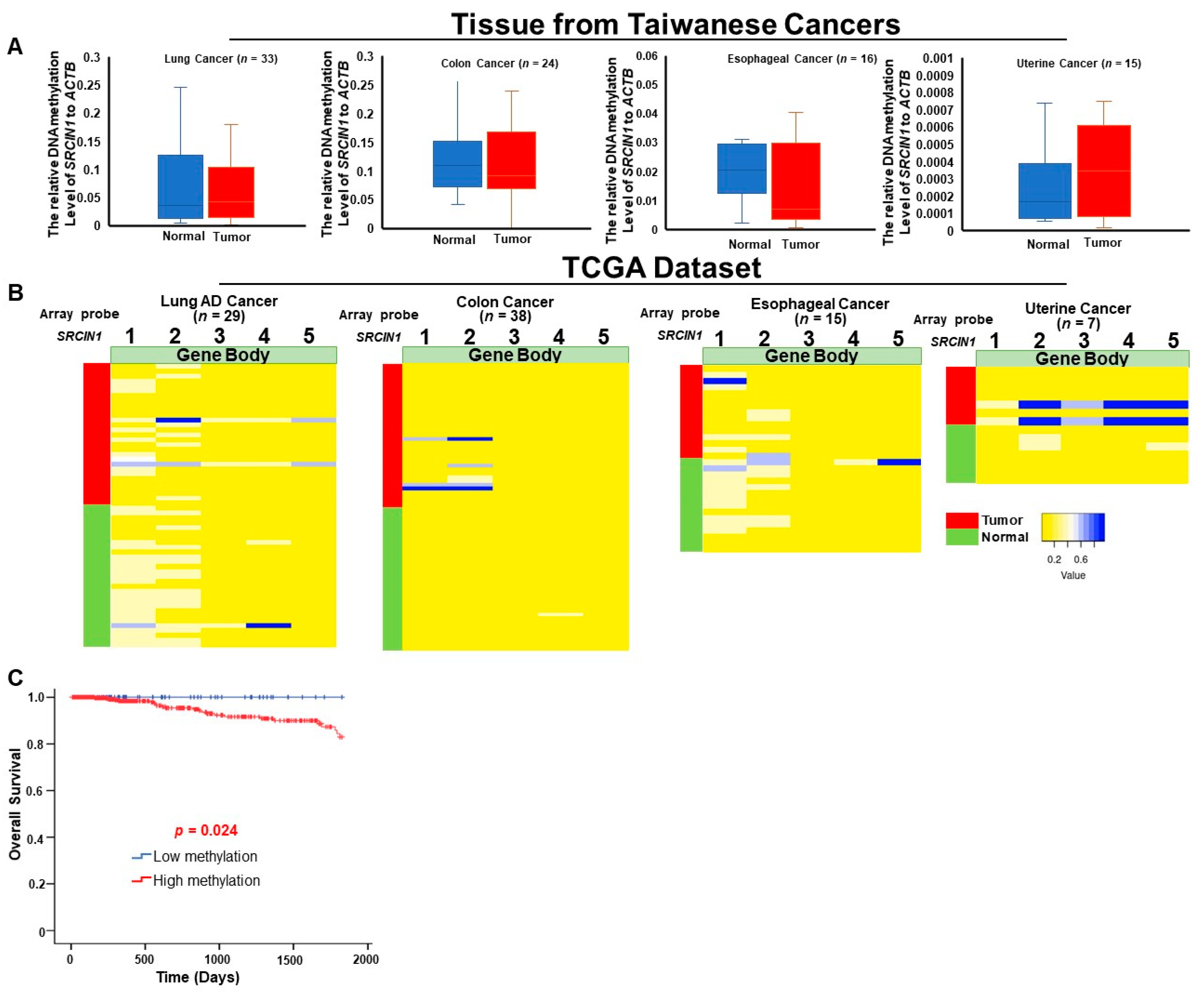
3.2. Hypermethylation of the SRCIN1 Gene Body and Elevated mRNA Expression in Taiwanese and TCGA Cohorts
3.3. Differential Methylation of the SRCIN1 Gene Is Specific to Breast Cancer and Are Correlated with Poor Survival
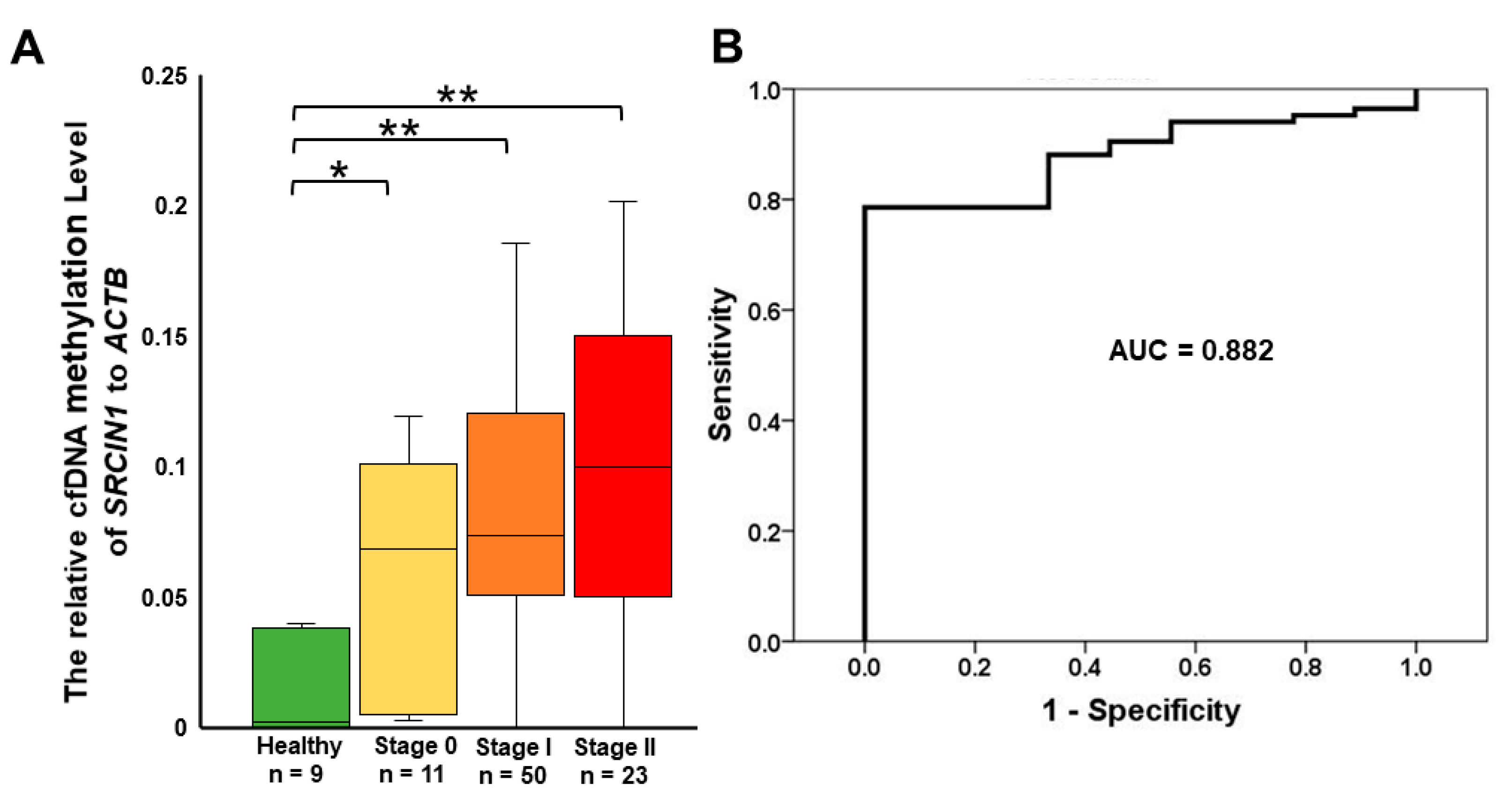
3.4. Detection of Circulating Methylated SRCIN1 in Breast Cancer Patients from Taiwan and the USA
3.5. SRCIN1 Expression Was Significantly Correlated with ER, PR, HER2 and TNBC Status and Cell Viability
3.6. SRCIN1 Silencing in the MCF-7 Cell Line Resulted in the Downregulation of the Estrogen Receptor, BCL2 and Cell Cyclin-Related Protein Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.M.; Parelukar, W. Breast cancers are rare diseases—And must be treated as such. NPJ Breast Cancer 2017, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Lenti, M.V.; Sottotetti, F.; Corazza, G.R. Tackling the clinical complexity of breast cancer. Drugs Context 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Breast cancer: A molecularly heterogenous disease needing subtype-specific treatments. Med. Sci. 2020, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Annaratone, L.; Cascardi, E.; Vissio, E.; Sarotto, I.; Chmielik, E.; Sapino, A.; Berrino, E.; Marchiò, C. The multifaceted nature of tumor microenvironment in breast carcinomas. Pathobiology 2020, 87, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Goldgar, S.; Heerboth, S.; Leary, M.; Housman, G.; Moulton, K.; Sarkar, S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014, 34, 1071–1077. [Google Scholar]
- Lamb, C.A.; Vanzulli, S.; Lanari, C.L.M. Hormone Receptors in Breast Cancer: More Than Estrogen Receptors. Medicina (B Aires) 2019, 79, 540–545. [Google Scholar] [PubMed]
- Chan, C.W.; Law, B.M.; So, W.K.; Chow, K.M.; Waye, M.M. Novel strategies on personalized medicine for breast cancer treatment: An update. Int. J. Mol. Sci. 2017, 18, 2423. [Google Scholar] [CrossRef]
- Dieterich, M.; Stubert, J.; Reimer, T.; Erickson, N.; Berling, A. Influence of lifestyle factors on breast cancer risk. Breast Care 2014, 9, 407–414. [Google Scholar] [CrossRef]
- Myers, E.R.; Moorman, P.; Gierisch, J.M.; Havrilesky, L.J.; Grimm, L.J.; Ghate, S.; Davidson, B.; Mongtomery, R.C.; Crowley, M.J.; McCrory, D.C.; et al. Benefits and Harms of Breast Cancer Screening: A Systematic Review. JAMA 2015, 314, 1615–1634. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Dershaw, D.D.; Kopans, D.; Evans, P.; Monsees, B.; Monticciolo, D.; Brenner, R.J.; Bassett, L.; Berg, W.; Feig, S.; et al. Breast Cancer Screening With Imaging: Recommendations From the Society of Breast Imaging and the ACR on the Use of Mammography, Breast MRI, Breast Ultrasound, and Other Technologies for the Detection of Clinically Occult Breast Cancer. J. Am. Coll. Radiol. 2010, 7, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Shelling, A.; Muthukaruppan, A.; Lasham, A.; Blenkiron, C.; Laking, G.; Print, C. Predictive and prognostic molecular markers for cancer medicine. Ther. Adv. Med. Oncol. 2010, 2, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.; Goldstein, L.J. Prognostic and predictive factors in early-stage breast cancer. Oncologist 2004, 9, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Schiano, C.; Soricelli, A.; De Nigris, F.; Napoli, C. New challenges in integrated diagnosis by imaging and osteo-immunology in bone lesions. Expert Rev. Clin. Immunol. 2019, 15, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; McDermott, E.W.; Crown, J. Blood-based biomarkers in breast cancer: From proteins to circulating tumor cells to circulating tumor DNA. Tumor Biol. 2018, 40, 1010428318776169. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann. Oncol. 2017, 28, 1117–1123. [Google Scholar] [CrossRef]
- Sher, G.; Salman, N.A.; Khan, A.Q.; Prabhu, K.S.; Raza, A.; Kulinski, M.; Dermime, S.; Haris, M.; Junejo, K.; Uddin, S. Epigenetic and breast cancer therapy: Promising diagnostic and therapeutic applications. Semin. Cancer Biol. 2022, 83, 152–165. [Google Scholar] [CrossRef]
- Bhat, S.A.; Majid, S.; Wani, H.A.; Rashid, S. Diagnostic utility of epigenetics in breast cancer—A review. Cancer Treat. Res. Commun. 2019, 19, 100125. [Google Scholar] [CrossRef]
- Avraham, A.; Cho, S.S.; Uhlmann, R.; Polak, M.L.; Sandbank, J.; Karni, T.; Pappo, I.; Halperin, R.; Vaknin, Z.; Sella, A.; et al. Tissue Specific DNA Methylation in Normal Human Breast Epithelium and in Breast Cancer. PLoS ONE 2014, 9, e91805. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, L.; Chen, J.; Su, J.; Shen, W.; Liu, B.; Zhou, J.; Yu, S.; Qian, J. A four-gene signature for prognosis in breast cancer patients with hypermethylated IL15RA. Oncol. Lett. 2019, 17, 4245–4254. [Google Scholar] [CrossRef] [PubMed]
- Neves Rebello Alves, L.; Dummer Meira, D.; Poppe Merigueti, L.; Correia Casotti, M.; do Prado Ventorim, D.; Ferreira Figueiredo Almeida, J.; Pereira de Sousa, V.; Cindra Sant’Ana, M.; Gonçalves Coutinho da Cruz, R.; Santos Louro, L. Biomarkers in breast cancer: An old story with a new end. Genes 2023, 14, 1364. [Google Scholar] [CrossRef]
- Seale, K.N.; Tkaczuk, K.H. Circulating biomarkers in breast cancer. Clin. Breast Cancer 2022, 22, e319–e331. [Google Scholar] [CrossRef] [PubMed]
- Buocikova, V.; Rios-Mondragon, I.; Pilalis, E.; Chatziioannou, A.; Miklikova, S.; Mego, M.; Pajuste, K.; Rucins, M.; Yamani, N.E.; Longhin, E.M. Epigenetics in breast cancer therapy—New strategies and future nanomedicine perspectives. Cancers 2020, 12, 3622. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Davidsson, S.; Fridfeldt, J.; Giunchi, F.; Fiano, V.; Grasso, C.; Zelic, R.; Richiardi, L.; Andrén, O.; Pettersson, A.; et al. Quantity and quality of nucleic acids extracted from archival formalin fixed paraffin embedded prostate biopsies. BMC Med. Res. Methodol. 2018, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Liao, L.-M.; Ansar, M.; Lin, S.-Y.; Hsu, W.-W.; Su, C.-M.; Chung, Y.-M.; Liu, C.-C.; Hung, C.-S.; Lin, R.-K. Automatic detection of the circulating cell-free methylated DNA pattern of GCM2, ITPRIPL1 and CCDC181 for detection of early breast cancer and surgical treatment response. Cancers 2021, 13, 1375. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-K.; Su, C.-M.; Lin, S.-Y.; Thi Anh Thu, L.; Liew, P.-L.; Chen, J.-Y.; Tzeng, H.-E.; Liu, Y.-R.; Chang, T.-H.; Lee, C.-Y. Hypermethylation of TMEM240 predicts poor hormone therapy response and disease progression in breast cancer. Mol. Med. 2022, 28, 67. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Liew, P.-L.; Ansar, M.; Lin, S.-Y.; Wang, S.-C.; Hung, C.-S.; Chen, J.-Y.; Jain, S.; Lin, R.-K. Hypermethylation and decreased expression of TMEM240 are potential early-onset biomarkers for colorectal cancer detection, poor prognosis, and early recurrence prediction. Clin. Epigenet. 2020, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef]
- Arai, E.; Sakamoto, H.; Ichikawa, H.; Totsuka, H.; Chiku, S.; Gotoh, M.; Mori, T.; Nakatani, T.; Ohnami, S.; Nakagawa, T.; et al. Multilayer-omics analysis of renal cell carcinoma, including the whole exome, methylome and transcriptome. Int. J. Cancer 2014, 135, 1330–1342. [Google Scholar] [CrossRef]
- Ansar, M.; Wang, C.-J.; Wang, Y.-H.; Shen, T.-H.; Hung, C.-S.; Chang, S.-C.; Lin, R.-K. SMAD3 hypomethylation as a biomarker for early prediction of colorectal cancer. Int. J. Mol. Sci. 2020, 21, 7395. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.A.; Shimbo, T.; Takaku, M.; Thomas, J.W.; Auerbach, S.; Bennett, B.D.; Bucher, J.R.; Burkholder, A.B.; Day, F.; Du, Y. DNA methylation in mice is influenced by genetics as well as sex and life experience. Nat. Commun. 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Puzanov, G.A.; Dashinimaev, E.B.; Vishnyakova, K.S.; Kondratieva, T.T.; Chegodaev, Y.S.; Postnov, A.Y.; Senchenko, V.N.; Yegorov, Y.E. Tumor Suppressor Properties of Small C-Terminal Domain Phosphatases in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 12986. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; LaPierre, M.P.; Gasser, E.; Denzler, R.; Yang, Y.; Rülicke, T.; Kero, J.; Latreille, M.; Stoffel, M. Loss of microRNA-7a2 induces hypogonadotropic hypogonadism and infertility. J. Clin. Investig. 2017, 127, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Beggs, A.D.; James, J.; Caldwell, G.; Prout, T.; Dilworth, M.P.; Taniere, P.; Iqbal, T.; Morton, D.G.; Matthews, G. Discovery and validation of methylation biomarkers for ulcerative colitis associated neoplasia. Inflamm. Bowel Dis. 2018, 24, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Huang, T.; Ye, G.; Wang, B.; Zhang, X. Methylation of SFRP2 gene as a promising noninvasive biomarker using feces in colorectal cancer diagnosis: A systematic meta-analysis. Sci. Rep. 2016, 6, 33339. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.-L.; Chen, M.-H.; Huang, K.-H.; Chang, S.-C.; Lin, C.-H.; Chao, Y.; Lo, S.-S.; Li, A.F.-Y.; Wu, C.-W.; Shyr, Y.-M. Analysis of the clinical significance of DNA methylation in gastric cancer based on a genome-wide high-resolution array. Clin. Epigenet. 2019, 11, 154. [Google Scholar] [CrossRef]
- Cai, T.-t.; Muhali, F.-s.; Song, R.-h.; Qin, Q.; Wang, X.; Shi, L.-f.; Jiang, W.-j.; Xiao, L.; Li, D.-f.; Zhang, J.-a. Genome-wide DNA methylation analysis in Graves’ disease. Genomics 2015, 105, 204–210. [Google Scholar] [CrossRef]
- Salgado, C.M.; Alaggio, R.; Ciolfi, A.; Zin, A.; Camassei, F.D.; Pedace, L.; Milano, G.M.; Serra, A.; Di Giannatale, A.; Mastronuzzi, A. Pediatric BCOR-altered tumors from soft tissue/kidney display specific DNA methylation profiles. Mod. Pathol. 2023, 36, 100039. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.K.; Mellor, P.; Smith, S.E.; Kendall, S.; Just, N.A.; Vizeacoumar, F.S.; Sarker, S.; Phillips, Z.; Alvi, R.; Saxena, A. Epigenetic silencing of CREB3L1 by DNA methylation is associated with high-grade metastatic breast cancers with poor prognosis and is prevalent in triple negative breast cancers. Breast Cancer Res. 2016, 18, 12. [Google Scholar] [CrossRef]
- Sun, F.; Lian, Y.; Wang, J.; Hu, L.; Luo, J.; Yu, J. KIF26B in the Prognosis and Immune Biomarking of Various Cancers: A Pan-Cancer Study. J. Oncol. 2022, 2022, 4829697. [Google Scholar] [CrossRef]
- Stastny, I.; Zubor, P.; Kajo, K.; Kubatka, P.; Golubnitschaja, O.; Dankova, Z. Aberrantly methylated cfDNA in body fluids as a promising diagnostic tool for early detection of breast cancer. Clin. Breast Cancer 2020, 20, e711–e722. [Google Scholar] [CrossRef]
- Loomans-Kropp, H.A.; Song, Y.; Gala, M.; Parikh, A.R.; Van Seventer, E.E.; Alvarez, R.; Hitchins, M.P.; Shoemaker, R.H.; Umar, A. Methylated Septin9 (m SEPT9): A Promising Blood-Based Biomarker for the Detection and Screening of Early-Onset Colorectal Cancer. Cancer Res. Commun. 2022, 2, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene body methylation in cancer: Molecular mechanisms and clinical applications. Clin Epigenet. 2022, 14, 154. [Google Scholar] [CrossRef]
- de Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef]
- Cappetta, M.; Fernandez, L.; Brignoni, L.; Artagaveytia, N.; Bonilla, C.; López, M.; Esteller, M.; Bertoni, B.; Berdasco, M. Discovery of novel DNA methylation biomarkers for non-invasive sporadic breast cancer detection in the Latino population. Mol. Oncol. 2021, 15, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Garcia-Closas, M.; Orr, N.; Fletcher, O.; Jones, M.; Ashworth, A.; Swerdlow, A.; Thorne, H.; Riboli, E.; Vineis, P. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012, 72, 2304–2313. [Google Scholar] [CrossRef]
- Guan, Z.; Yu, H.; Cuk, K.; Zhang, Y.; Brenner, H. Whole-blood DNA methylation markers in early detection of breast cancer: A systematic literature review. Cancer Epidemiol. Biomark. Prev. 2019, 28, 496–505. [Google Scholar] [CrossRef]
- Iwamoto, T.; Yamamoto, N.; Taguchi, T.; Tamaki, Y.; Noguchi, S. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res. Treat. 2011, 129, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Stöcker, S.; Schott, S.; Heil, J.; Marme, F.; Cuk, K.; Chen, B.; Golatta, M.; Zhou, Y.; Sutter, C. The association between breast cancer and S100P methylation in peripheral blood by multicenter case–control studies. Carcinogenesis 2017, 38, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; LeProust, E.M.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.; Clynes, M.; Doolan, P.; Mehta, J.; Rani, S.; Crown, J.; O’driscoll, L. SNIP/p140Cap mRNA expression is an unfavourable prognostic factor in breast cancer and is not expressed in normal breast tissue. Br. J. Cancer 2008, 98, 1641–1645. [Google Scholar] [CrossRef]
- Grasso, S.; Chapelle, J.; Salemme, V.; Aramu, S.; Russo, I.; Vitale, N.; di Cantogno, L.V.; Dallaglio, K.; Castellano, I.; Amici, A. The scaffold protein p140Cap limits ERBB2-mediated breast cancer progression interfering with Rac GTPase-controlled circuitries (vol 8, 14797, 2017). Nat. Commun. 2018, 9, 14797. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Yu, S.; Zhuang, Y.; Zhang, Q.; Wang, J.; Gao, X. SRCIN1 regulated by circCCDC66/miR-211 Is upregulated and promotes cell proliferation in non-small-cell lung cancer. BioMed Res. Int. 2020, 2020, 5307641. [Google Scholar] [CrossRef] [PubMed]
- Salemme, V.; Vedelago, M.; Sarcinella, A.; Moietta, F.; Piccolantonio, A.; Moiso, E.; Centonze, G.; Manco, M.; Guala, A.; Lamolinara, A.; et al. p140Cap inhibits β-Catenin in the breast cancer stem cell compartment instructing a protective anti-tumor immune response. Nat. Commun. 2023, 14, 2350. [Google Scholar] [CrossRef]
- Di Stefano, P.; Cabodi, S.; Boeri Erba, E.; Margaria, V.; Bergatto, E.; Giuffrida, M.G.; Silengo, L.; Tarone, G.; Turco, E.; Defilippi, P. P130Cas-associated protein (p140Cap) as a new tyrosine-phosphorylated protein involved in cell spreading. Mol. Biol. Cell 2004, 15, 787–800. [Google Scholar] [CrossRef]
- Lee, A.V.; Oesterreich, S.; Davidson, N.E. MCF-7 cells—Changing the course of breast cancer research and care for 45 years. J. Natl. Cancer Inst. 2015, 107, djv073. [Google Scholar] [CrossRef] [PubMed]
- Eom, Y.H.; Kim, H.S.; Lee, A.; Song, B.J.; Chae, B.J. BCL2 as a subtype-specific prognostic marker for breast cancer. J. Breast Cancer 2016, 19, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed]
- Michalides, R.; Van Tinteren, H.; Balkenende, A.; Vermorken, J.; Benraadt, J.; Huldij, J.; Van Diest, P. Cyclin A is a prognostic indicator in early stage breast cancer with and without tamoxifen treatment. Br. J. Cancer 2002, 86, 402–408. [Google Scholar] [CrossRef]
- Loukil, A.; Cheung, C.T.; Bendris, N.; Lemmers, B.; Peter, M.; Blanchard, J.M. Cyclin A2: At the crossroads of cell cycle and cell invasion. World J. Biol. Chem. 2015, 6, 346. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total b (%) | Methylation c | p Value | Total (%) | Expression d | p Value | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Low (%) | High (%) | Low (%) | Normal (%) | High (%) | ||||||
| Overall | 102 | 39 (38.2) | 63 (61.7) | 43 | 4 (9.3) | 18 (41.8) | 21 (48.8) | |||
| Stage | 96 | 36 (37.5) | 60 (62.5) | 0.427 | 41 | 4 (9.8) | 16 (39.0) | 21 (51.2) | 0.227 | |
| I and II | 69 (71.9) | 25 (36.2) | 44 (63.8) | 33 (80.5) | 3 (9.1) | 15 (45.5) | 15 (45.5) | |||
| III and IV | 27 (28.1) | 11 (40.7) | 16 (59.3) | 8 (19.5) | 1 (12.5) | 1 (12.5) | 6 (75.0) | |||
| Histological Type | 98 | 37 (37.8) | 61 (62.2) | 0.385 | 42 | 4 (9.5) | 17 (40.5) | 21 (50.0) | 0.471 | |
| IDC | 96 (98) | 37 (38.5) | 59 (61.5) | 41 (97.6) | 4 (9.8) | 16 (39.0) | 21 (51.2) | |||
| ILC | 2 (2) | 0 (0) | 2 (100) | 1 (2.4) | 0 (0.0) | 1 (100) | 0 (0) | |||
| Tumor size | 97 | 36 (37.1) | 62 (62.9) | 0.089 | 41 | 4 (9.5) | 17 (40.5) | 20 (50.0) | 0.830 | |
| T0–T1 | 25 (25.8) | 6 (24.0) | 19 (76.0) | 16 (39.0) | 2 (12.5) | 7 (43.8) | 7 (43.8) | |||
| T2–T4 | 72 (74.2) | 30 (41.7) | 42 (58.3) | 25 (61.0) | 2 (8.0) | 10 (40.0) | 13 (61.0) | |||
| Lymph node involvement | 94 | 34 (36.2) | 60 (63.8) | 0.507 | 40 | 4 (10.0) | 17 (42.7) | 19 (47.5) | 0.567 | |
| N0 | 40 (42.6) | 14 (41.2) | 26 (65.0) | 22 (55.0) | 2 (9.1) | 11 (50.0) | 9 (40.9) | |||
| N1–N3 | 54 (57.4) | 20 (37.0) | 34 (63.0) | 18 (45.0) | 2 (11.1) | 6 (33.3) | 10 (55.6) | |||
| Subtypes | 0.352 | 0.935 | ||||||||
| Luminal A | 15 (17.4) | 8 (53.3) | 7 (46.7) | 5 (17.2) | 0 (0.0) | 2 (40.0) | 3 (60.0) | |||
| Luminal B | 37 (43.0) | 13 (35.1) | 24 (64.9) | 11 (37.9) | 1 (9.1) | 4 (36.4) | 6 (54.5) | |||
| HER2 | 18 (20.9) | 5 (27.8) | 13 (72.2) | 7 (24.1) | 0 (0.0) | 3 (42.9) | 4 (57.1) | |||
| TNBC | 16 (18.6) | 8 (50.0) | 8 (50.0) | 6 (20.1) | 0 (0.0) | 2 (33.3) | 4 (66.7) | |||
| Tumor markers | ||||||||||
| Estrogen | 93 | 37 (39.8) | 56 (60.2) | 0.356 | 38 | 2 (5.3) | 16 (42.1) | 20 (52.6) | 0.497 | |
| Positive | 62 (66.7) | 26 (41.9) | 36 (58.1) | 25 (65.8) | 2 (8.0) | 11 (44.0) | 12 (48.0) | |||
| Negative | 31 (33.3) | 11 (35.5) | 20 (64.5) | 13 (34.2) | 0 (0.0) | 5 (38.5) | 8 (61.5) | |||
| Progesterone | 93 | 37 (39.8) | 56 (60.2) | 0.465 | 38 | 2 (5.3) | 16 (42.1) | 20 (52.6) | 0.762 | |
| Positive | 51 (54.8) | 21 (41.5) | 30 (58.8) | 24 (63.2) | 1 (4.2) | 10 (41.7) | 13 (54.2) | |||
| Negative | 42 (45.2) | 16 (43.2) | 26 (61.9) | 14 (36.8) | 1 (7.1) | 6 (42.9) | 7 (50.0) | |||
| HER2 | 92 | 36 (39.1) | 56 (60.9) | 0.094 | 37 | 1 (2.7) | 16 (43.2) | 20 (54.1) | 0.542 | |
| Positive | 55 (59.8) | 18 (32.7) | 37 (67.3) | 18 (48.6) | 1 (5.6) | 7 (38.9) | 10 (55.6) | |||
| Negative | 37 (40.2) | 18 (48.6) | 19 (51.4) | 19 (51.4) | 0 (0.0) | 9 (47.4) | 10 (52.6) | |||
| Ki-67 | 84 | 33 (39.3) | 51 (60.7) | 0.441 | 30 | 2 (6.7) | 15 (50.0) | 13 (43.3) | 0.725 | |
| >14% | 59 (70.2) | 24 (40.7) | 35 (59.3) | 22 (73.3) | 1 (4.5) | 11 (50.0) | 10 (45.5) | |||
| <14% | 25 (29.8) | 9 (36.0) | 116 (64.0) | 8 (26.7) | 1 (12.5) | 4 (50.0) | 3 (26.7) | |||
| SRCIN1 Methylation | |||
|---|---|---|---|
| Total N | Low N (%) | High b N (%) | |
| 93 | 27 (29.0) | 66 (71.0) | |
| Healthy | 9 | 9 (100.0) | 0 (0.0) |
| Stage 0 | 11 | 4 (36.4) | 7 (63.6) |
| Stage I | 50 | 10 (20.0) | 40 (80.0) |
| Stage II | 23 | 4 (17.4) | 19 (82.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, H.-T.; Hung, C.-S.; Davis, C.; Su, C.-M.; Liao, L.-M.; Shih, H.-M.; Lee, K.-D.; Ansar, M.; Lin, R.-K. Hypermethylation of the Gene Body in SRCIN1 Is Involved in Breast Cancer Cell Proliferation and Is a Potential Blood-Based Biomarker for Early Detection and a Poor Prognosis. Biomolecules 2024, 14, 571. https://doi.org/10.3390/biom14050571
Shen H-T, Hung C-S, Davis C, Su C-M, Liao L-M, Shih H-M, Lee K-D, Ansar M, Lin R-K. Hypermethylation of the Gene Body in SRCIN1 Is Involved in Breast Cancer Cell Proliferation and Is a Potential Blood-Based Biomarker for Early Detection and a Poor Prognosis. Biomolecules. 2024; 14(5):571. https://doi.org/10.3390/biom14050571
Chicago/Turabian StyleShen, Hsieh-Tsung, Chin-Sheng Hung, Clilia Davis, Chih-Ming Su, Li-Min Liao, Hsiu-Ming Shih, Kuan-Der Lee, Muhamad Ansar, and Ruo-Kai Lin. 2024. "Hypermethylation of the Gene Body in SRCIN1 Is Involved in Breast Cancer Cell Proliferation and Is a Potential Blood-Based Biomarker for Early Detection and a Poor Prognosis" Biomolecules 14, no. 5: 571. https://doi.org/10.3390/biom14050571