
Fusion with ARRDC1 or CD63: A Strategy to Enhance p53 Loading into Extracellular Vesicles for Tumor Suppression
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Cell Culture and Transfection
2.3. sEV Purification
2.4. Biophysical Characterization and Imaging
2.5. Immunoblot Analysis and Antibodies
2.6. Immunofluorescence Labeling and Confocal Imaging
2.7. Labeling sEVs and Tracking Cellular Uptake
2.8. Real-Time Quantitative Polymerase Chain Reaction
2.9. Cell Viability Assay
2.10. Apoptosis Detection
2.11. Statistical Analysis
3. Results
3.1. Expression, Function, and Localization of p53 Fusion Proteins
3.2. ARRDC1 and CD63 Enhance ARMMs and Exosome Gene Expression
3.3. Improved sEV Production and p53 Loading Efficiency by ARP or CDP Transfection
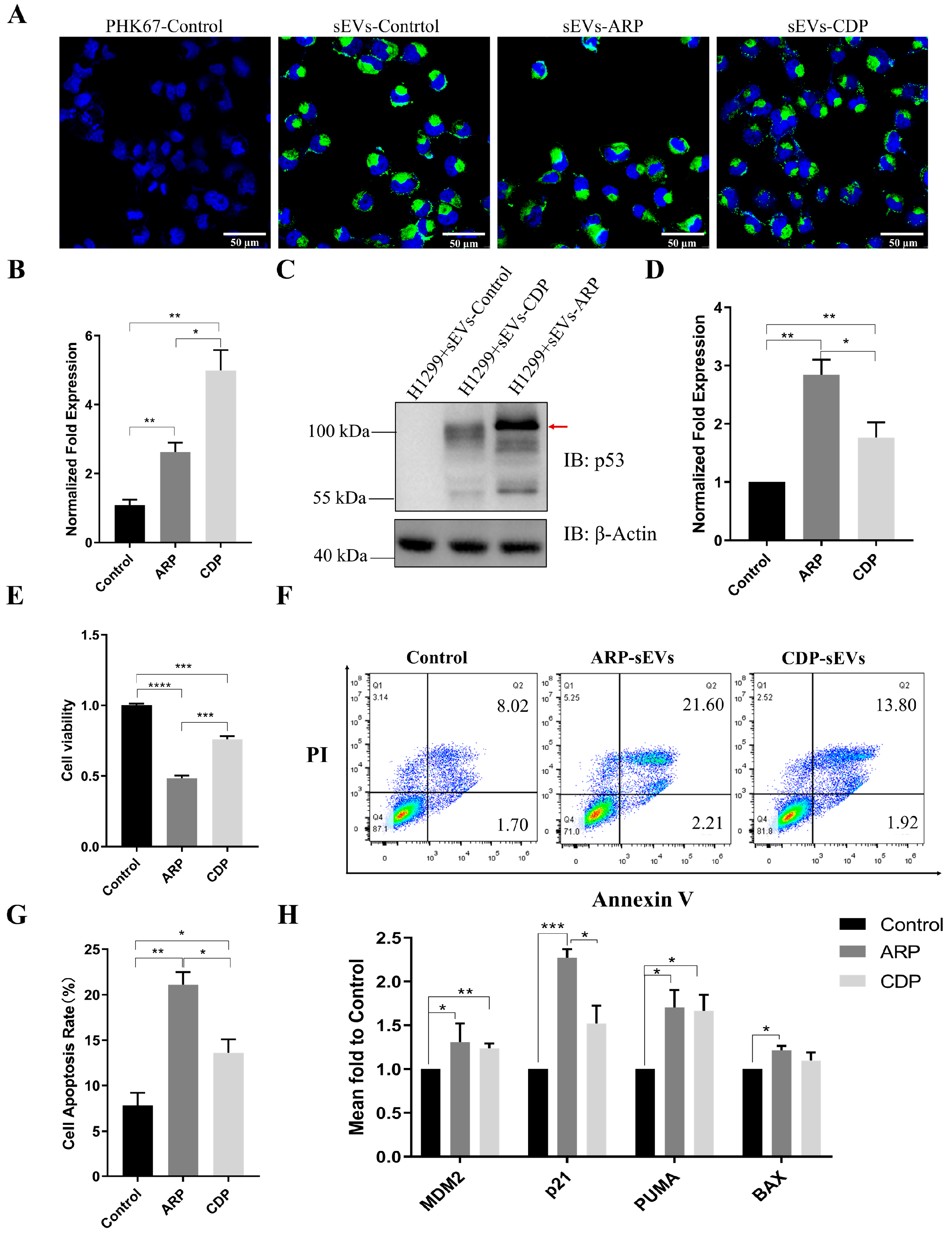
3.4. ARP-sEVs Outperform CDP-sEVs in Anti-Tumor Effects on H1299 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA Delivery by Extracellular Vesicles in Mammalian Cells and Its Applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtiö, J.; El Andaloussi, S. Cells Release Subpopulations of Exosomes with Distinct Molecular and Biological Properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and Release of Arrestin Domain-Containing Protein 1-Mediated Microvesicles (ARMMs) at Plasma Membrane by Recruitment of TSG101 Protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes Facilitate Therapeutic Targeting of Oncogenic KRAS in Pancreatic Cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Couch, Y.; Buzàs, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Théry, C.; et al. A Brief History of Nearly EV-Erything—The Rise and Rise of Extracellular Vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zheng, S.; Luo, Y.; Wang, B. Exosome Theranostics: Biology and Translational Medicine. Theranostics 2018, 8, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A. Tumor Microenvironment Derived Exosomes Pleiotropically Modulate Cancer Cell Metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-Inflammatory Drugs from the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Cianciaruso, C.; Hansen, S.K.; De Palma, M. EVIR: Chimeric Receptors That Enhance Dendritic Cell Cross-Dressing with Tumor Antigens. Nat. Methods 2018, 15, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Gangadaran, P.; Ahn, B.-C. Extracellular Vesicle-and Extracellular Vesicle Mimetics-Based Drug Delivery Systems: New Perspectives, Challenges, and Clinical Developments. Pharmaceutics 2020, 12, 442. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-Mediated Production of Exosomes during Hypoxia Is Protective in Renal Tubular Cells. Am. J. Physiol. Physiol. 2017, 313, F906–F913. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of SiRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Nkosi, D.; Conlon, M.M.; York, S.B.; Liu, X.; Tremblay, D.C.; Meckes, D.G., Jr. CD63 Regulates Epstein-Barr Virus LMP1 Exosomal Packaging, Enhancement of Vesicle Production, and Noncanonical NF-ΚB Signaling. J. Virol. 2017, 91, e02251-16. [Google Scholar] [CrossRef]
- Yao, X.; Lyu, P.; Yoo, K.; Yadav, M.K.; Singh, R.; Atala, A.; Lu, B. Engineered Extracellular Vesicles as Versatile Ribonucleoprotein Delivery Vehicles for Efficient and Safe CRISPR Genome Editing. J. Extracell. Vesicles 2021, 10, e12076. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a Versatile Platform for Intracellular Delivery of Macromolecules. Nat. Commun. 2018, 9, 960. [Google Scholar] [CrossRef] [PubMed]
- Keysberg, C.; Schneider, H.; Otte, K. Production Cell Analysis and Compound-Based Boosting of Small Extracellular Vesicle Secretion Using a Generic and Scalable Production Platform. Biotechnol. Bioeng. 2022, 120, 987–999. [Google Scholar] [CrossRef]
- Song, Z.; Mao, J.; Barrero, R.A.; Wang, P.; Zhang, F.; Wang, T. Development of a CD63 Aptamer for Efficient Cancer Immunochemistry and Immunoaffinity-Based Exosome Isolation. Molecules 2020, 25, 5585. [Google Scholar] [CrossRef]
- Tan, S.S.; Yin, Y.; Lee, T.; Lai, R.C.; Yeo, R.W.Y.; Zhang, B.; Choo, A.; Lim, S.K. Therapeutic MSC Exosomes Are Derived from Lipid Raft Microdomains in the Plasma Membrane. J. Extracell. Vesicles 2013, 2, 22614. [Google Scholar] [CrossRef]
- Zhang, R.; Bu, T.; Cao, R.; Li, Z.; Wang, C.; Huang, B.; Wei, M.; Yuan, L.; Yang, G. An Optimized Exosome Production Strategy for Enhanced Yield While without Sacrificing Cargo Loading Efficiency. J. Nanobiotechnology 2022, 20, 463. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and Challenges of Extracellular Vesicle-Based Drug Delivery System: Considering Cell Source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Jafari, D.; Malih, S.; Eini, M.; Jafari, R.; Gholipourmalekabadi, M.; Sadeghizadeh, M.; Samadikuchaksaraei, A. Improvement, Scaling-up, and Downstream Analysis of Exosome Production. Crit. Rev. Biotechnol. 2020, 40, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered Exosomes for Targeted Co-Delivery of MiR-21 Inhibitor and Chemotherapeutics to Reverse Drug Resistance in Colon Cancer. J. Nanobiotechnol. 2020, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Liang, X.; Pavlova, S.; Wiklander, O.P.B.; Corso, G.; Zhao, Y.; Saher, O.; Bost, J.; Zickler, A.M.; Piffko, A.; et al. Quantification of Extracellular Vesicles In Vitro and In Vivo Using Sensitive Bioluminescence Imaging. J. Extracell. Vesicles 2020, 9, 1800222. [Google Scholar] [CrossRef] [PubMed]
- Wallis, B.; Bowman, K.R.; Lu, P.; Lim, C.S. The Challenges and Prospects of P53-Based Therapies in Ovarian Cancer. Biomolecules 2023, 13, 159. [Google Scholar] [CrossRef]
- Liu, B.; Chen, F.; Wu, Y.; Wang, X.; Feng, M.; Li, Z.; Zhou, M.; Wang, Y.; Wu, L.; Liu, X.; et al. Enhanced Tumor Growth Inhibition by Mesenchymal Stem Cells Derived from IPSCs with Targeted Integration of Interleukin24 into RDNA Loci. Oncotarget 2017, 8, 40791–40803. [Google Scholar] [CrossRef]
- Wei, H.; Chen, Q.; Lin, L.; Sha, C.; Li, T.; Liu, Y.; Yin, X.; Xu, Y.; Chen, L.; Gao, W.; et al. Regulation of Exosome Production and Cargo Sorting. Int. J. Biol. Sci. 2020, 17, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT Machinery in Endosomal Sorting of Ubiquitylated Membrane Proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Ho, M.; Chen, T.; Liu, J.; Dowling, P.; Hideshima, T.; Zhang, L.; Morelli, E.; Camci-Unal, G.; Wu, X.; Tai, Y.-T. Targeting Histone Deacetylase 3 (HDAC3) in the Bone Marrow Microenvironment Inhibits Multiple Myeloma Proliferation by Modulating Exosomes and IL-6 Trans-Signaling. Leukemia 2020, 34, 196–209. [Google Scholar] [CrossRef]
- Mathieu, M.; Névo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D.; et al. Specificities of Exosome versus Small Ectosome Secretion Revealed by Live Intracellular Tracking of CD63 and CD9. Nat. Commun. 2021, 12, 4389. [Google Scholar] [CrossRef]
- Nam, E.; Lee, Y.-B.; Moon, C.; Chang, K.-A. Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2020, 21, 5007. [Google Scholar] [CrossRef] [PubMed]
- McConnell, R.E.; Higginbotham, J.N.; Shifrin Jr, D.A.; Tabb, D.L.; Coffey, R.J.; Tyska, M.J. The Enterocyte Microvillus is a Vesicle-Generating Organelle. J. Cell Biol. 2009, 185, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E. Syndecan–Syntenin–ALIX Regulates the Biogenesis of Exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, Q. Plasma Membrane-Derived Extracellular Microvesicles Mediate Non-Canonical Intercellular NOTCH Signaling. Nat. Commun. 2017, 8, 709. [Google Scholar] [CrossRef]
- Yang, J.; Wu, S.; Hou, L.; Zhu, D.; Yin, S.; Yang, G.; Wang, Y. Therapeutic Effects of Simultaneous Delivery of Nerve Growth Factor MRNA and Protein via Exosomes on Cerebral Ischemia. Mol. Ther.—Nucleic Acids 2020, 21, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, J.; Li, D.; Hao, L.; Li, Y.; Yi, D.; Yeung, K.W.K.; Chen, D.; Lu, W.W.; Pan, H.; et al. Engineering Stem Cells to Produce Exosomes with Enhanced Bone Regeneration Effects: An Alternative Strategy for Gene Therapy. J. Nanobiotechnology 2022, 20, 135. [Google Scholar] [CrossRef]
- Villamizar, O.; Waters, S.A.; Scott, T.; Grepo, N.; Jaffe, A.; Morris, K. V Mesenchymal Stem Cell Exosome Delivered Zinc Finger Protein Activation of Cystic Fibrosis Transmembrane Conductance Regulator. J. Extracell. Vesicles 2021, 10, e12053. [Google Scholar] [CrossRef]
- Shao, J.; Lu, J.; Zhu, W.; Yu, H.; Jing, X.; Wang, Y.-L.; Wang, X.; Wang, X.-J. Derepression of LOXL4 Inhibits Liver Cancer Growth by Reactivating Compromised P53. Cell Death Differ. 2019, 26, 2237–2252. [Google Scholar] [CrossRef]
- Carmo, L.A.S.; Bonjour, K.; Ueki, S.; Neves, J.S.; Liu, L.; Spencer, L.A.; Dvorak, A.M.; Weller, P.F.; Melo, R.C.N. CD63 is Tightly Associated with Intracellular, Secretory Events Chaperoning Piecemeal Degranulation and Compound Exocytosis in Human Eosinophils. J. Leucoc. Biol. 2016, 100, 391–401. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Zhang, Y.; He, J.; Liu, W.; Li, Z.; Zhang, Y.; Gu, A.; Zhao, M.; Liu, M.; Liu, X. Fusion with ARRDC1 or CD63: A Strategy to Enhance p53 Loading into Extracellular Vesicles for Tumor Suppression. Biomolecules 2024, 14, 591. https://doi.org/10.3390/biom14050591
Liu M, Zhang Y, He J, Liu W, Li Z, Zhang Y, Gu A, Zhao M, Liu M, Liu X. Fusion with ARRDC1 or CD63: A Strategy to Enhance p53 Loading into Extracellular Vesicles for Tumor Suppression. Biomolecules. 2024; 14(5):591. https://doi.org/10.3390/biom14050591
Chicago/Turabian StyleLiu, Min, Yu Zhang, Jianfeng He, Wanxi Liu, Zhexuan Li, Yiti Zhang, Ao Gu, Mingri Zhao, Mujun Liu, and Xionghao Liu. 2024. "Fusion with ARRDC1 or CD63: A Strategy to Enhance p53 Loading into Extracellular Vesicles for Tumor Suppression" Biomolecules 14, no. 5: 591. https://doi.org/10.3390/biom14050591