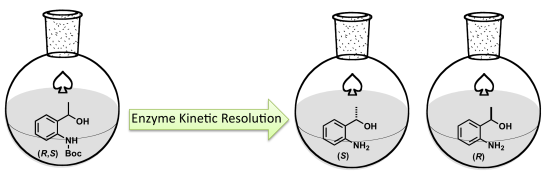
1. Introduction
Selenium- and tellurium-containing compounds have drawn the attention of the scientific community due to their biological properties [
1,
2,
3,
4]. Notwithstanding the intense activity in the field of selenium and tellurium chemistry over the last three decades, organometallic reagents are commonly employed on the preparation of organo-selenium and -tellurium compounds. Moreover, hypervalent organoselenium(IV) compounds (organoselenanes) and organotellurium(IV) compounds (organotelluranes) have been investigated as cysteine protease [
5,
6,
7,
8], protein tyrosine phosphatase [
9] and poliovirus 3C proteinase inhibitors [
10]. Considering the biological activities of organoselenanes and telluranes, we have described chemoenzymatic methodologies to synthesize selenium compounds without employing organolithium or organomagnesium reagents [
11,
12]. Herein, we report the preparation of enantiopure organochalcogenane precursors, (
R)- and (
S)-
tert-butyl 2-(1-hydroxyethyl)phenylcarbamate, employing enzymatic kinetic resolution (EKR) catalyzed by lipases. The chiral building blocks [(
R)-
I and (
S)-
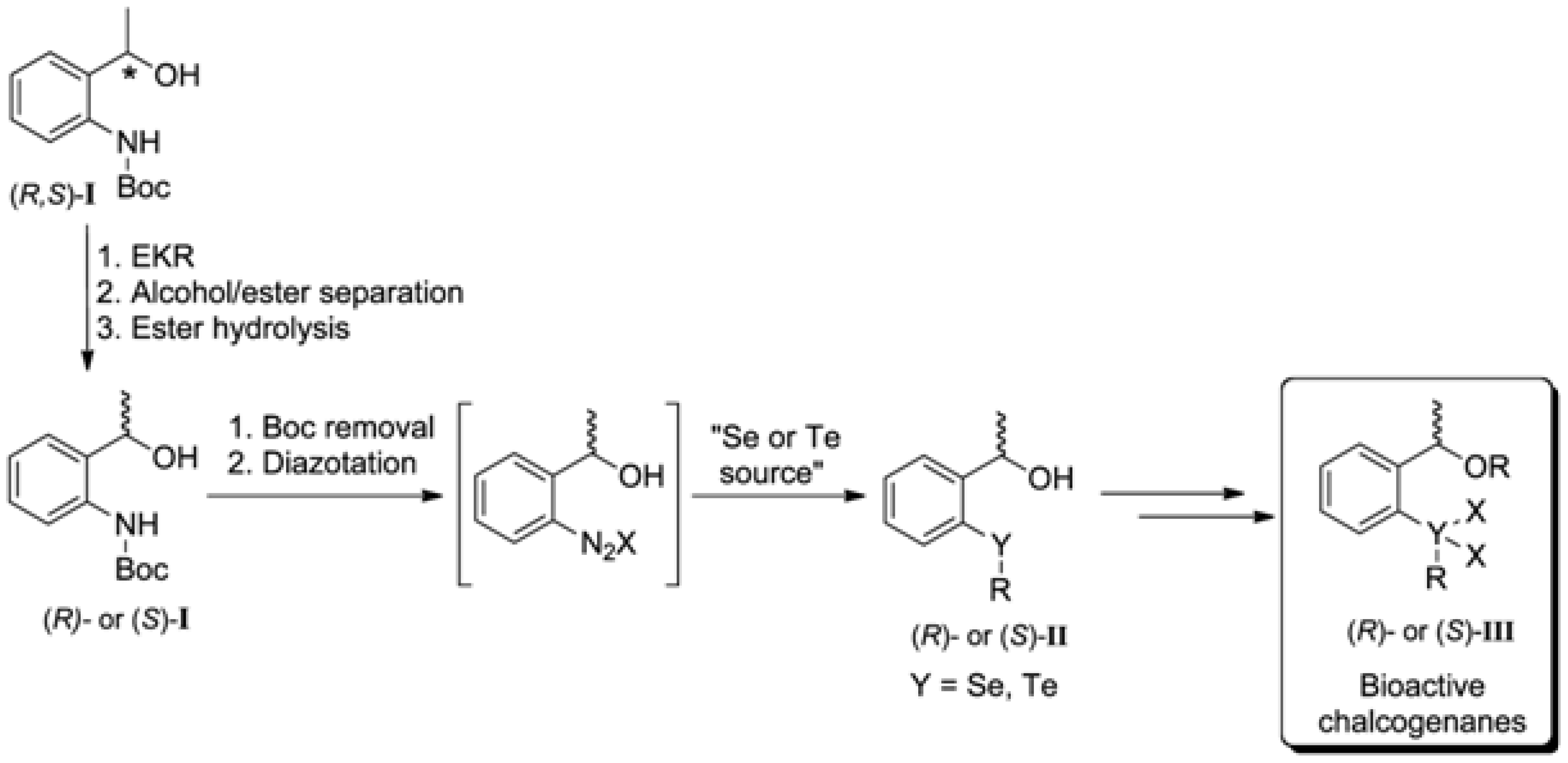
I] could be applied as advanced synthetic intermediates of organotelluranes and organoselenanes
III, containing an asymmetric center (
Scheme 1). It is possible to transform (
R)-
I and (
S)-
I into their respective arene diazonium salts, followed by a reaction with a nucleophilic selenium/tellurium specie to give selenides/tellurides
II, direct precursors of selenanes and telullaranes [
12].
Scheme 1.
Synthetic route to bioactive chalcogenanes [
5,
6,
9,
12].
Scheme 1.
Synthetic route to bioactive chalcogenanes [
5,
6,
9,
12].
3. Experimental Section
Commercially available materials were used without further purification. Lipase from Candida antarctica (fraction B, CAL-B) immobilized, and commercially available as Novozym® 435 was kindly donated by Novozymes Latin America Ltda. All solvents were HPLC or ACS grade. Solvents used for moisture sensitive operations were distilled from drying reagents under a nitrogen atmosphere: THF was distilled from Na/benzophenone.
Analytical thin-layer chromatography (TLC) was performed using aluminum-backed silica plates coated with a 0.25 mm thickness of silica gel 60 F254 (Merck), visualized with an ultraviolet light (l = 254 nm), followed by exposure to p-anisaldehyde solution or vanillin solution and heating. Standard chromatographic purification methods were followed using 35–70 mm (240–400 mesh) silica gel purchased from Acros Organics®.
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AC 200 spectrometer at operating frequencies of 200 (1H-NMR) and 50 MHz (13C-NMR). The 1H-NMR chemical shifts are reported in ppm relative to TMS peak. The data are reported as follows: chemical shift (δ), multiplicity (s = singlet, d = doublet, t = triplet, qd = quadruplet, dd = double dublet, td = triple dublet, m = multiplet), and coupling constant (J) in Hertz and integrated intensity. The 13C-NMR chemical shifts are reported in ppm relative to CDCl3 signal.
Reaction products were analyzed by a Shimadzu model GC-17A (FID) gas chromatograph equipped with a J&W Scientific HP5 column (30 m × 0.25 mm I.D.; 0.25 µm). The chromatographic conditions were as follows: Oven temperature initiated at 50 °C and increased at 10 °C/min; run time 20 min; injector temperature 230 °C; detector temperature 250 °C; injector split ratio 1:20; hydrogen carrier gas at a pressure of 100 kPa. The enantiomeric excesses of the products were determined by HPLC analyses performed in a Shimadzu model SPD-10Av instrument with UV-Vis detector (deuterium lamp 190–600 nm) and equipped with a Chiralcel® OD-H column (25 cm × 0.46 cm I.D.; Daicel Chemical Ind.) eluted with n-hexane (60%) and 2-propanol (99:1).
High-resolution mass spectra (HRMS) were acquired using a Bruker Daltonics MicroTOF instrument, operating in the electrospray ionization (ESI) mode.
Infrared spectra were recorded from KBr discs or from a thin film between NaCl plates on FTIR spectrometer (Bomem Michelson model 101). Absorption maxima (νmax) are reported in wavenumbers (cm−1).
Optical rotations were measured on a Perkin Elmer-343 digital polarimeter in a 1 mL cuvette with a 1 dm pathlength. All values are reported in the following format: [α]D(temperature of measurement) = specific rotation (concentration of the solution reported in units of 10 mg sample per 1 mL solvent used).
3.1. Synthesis of tert-butyl (2-Acetylphenyl)carbamate (2) (Adapted from References [13,14])
To a solution of the 1-(2-aminophenyl)ethanone (1, 1.35 g, 10 mmol) in anhydrous THF (100 mL) Boc)2O (6.48 g, 30 mmol) was added, followed by DMAP (122 mg, 1 mmol). The solution was stirred under reflux for 12 h then concentrated to dryness and partitioned between 0.5 mol L−1 HCl (100 mL) and EtOAc (100 mL). The aqueous layer was extracted with EtOAc (2 × 100 mL) and the combined organic phases were washed with brine (50 mL), dried over MgSO4, filtered and concentrated to afford the crude tert-butyl (2-acetylphenyl)carbamate (2) and the di-Boc derivative products. These compounds were separated by flash silica gel column chromatography eluted with hexane/EtOAc 9:1. (2-Acetylphenyl)carbamate (2) and the di-Boc derivative were isolated in 45% and 31% yields, respectively. The di-Boc compound (1.00 g) was dissolved in CH2Cl2 (100 mL) and Amberlyst 15 resin (1.00 g) was added. The mixture was stirred for 24 h in an orbital shaker. Then, the solvent was removed and the residue filtered through a silica gel column with hexane/EtOAc 9:1. The compound 2 was obtained in 67% yield. 1H-NMR (200 MHz, CDCl3); d (ppm): 10.95 (s, 1H); 8.46 (d, 1H, J = 8.3 Hz); 7.84 (dd, 1H, JA = 7.9 Hz; JB = 1.32 Hz); 7.50 (td, 1H, JA = 8.3 Hz; JB = 1.3 Hz); 7.01 (t, 1H, J = 7.9 Hz); 2.64 (s, 3H); 1.53 (s, 9H). 13C-NMR (50 MHz, CDCl3); d (ppm): 202.5; 153.4; 142.0; 135.2; 131.9; 121.6; 121.2; 119.4; 80.7; 28.8. IV (KBr), cm−1: 3432; 2947; 1713; 1656; 1562; 1239; 1108; 739. HRMS (ESI), [M+Na]+: Calculated for C13H17NO3Na: 258.1106. Found: 258, 1104.
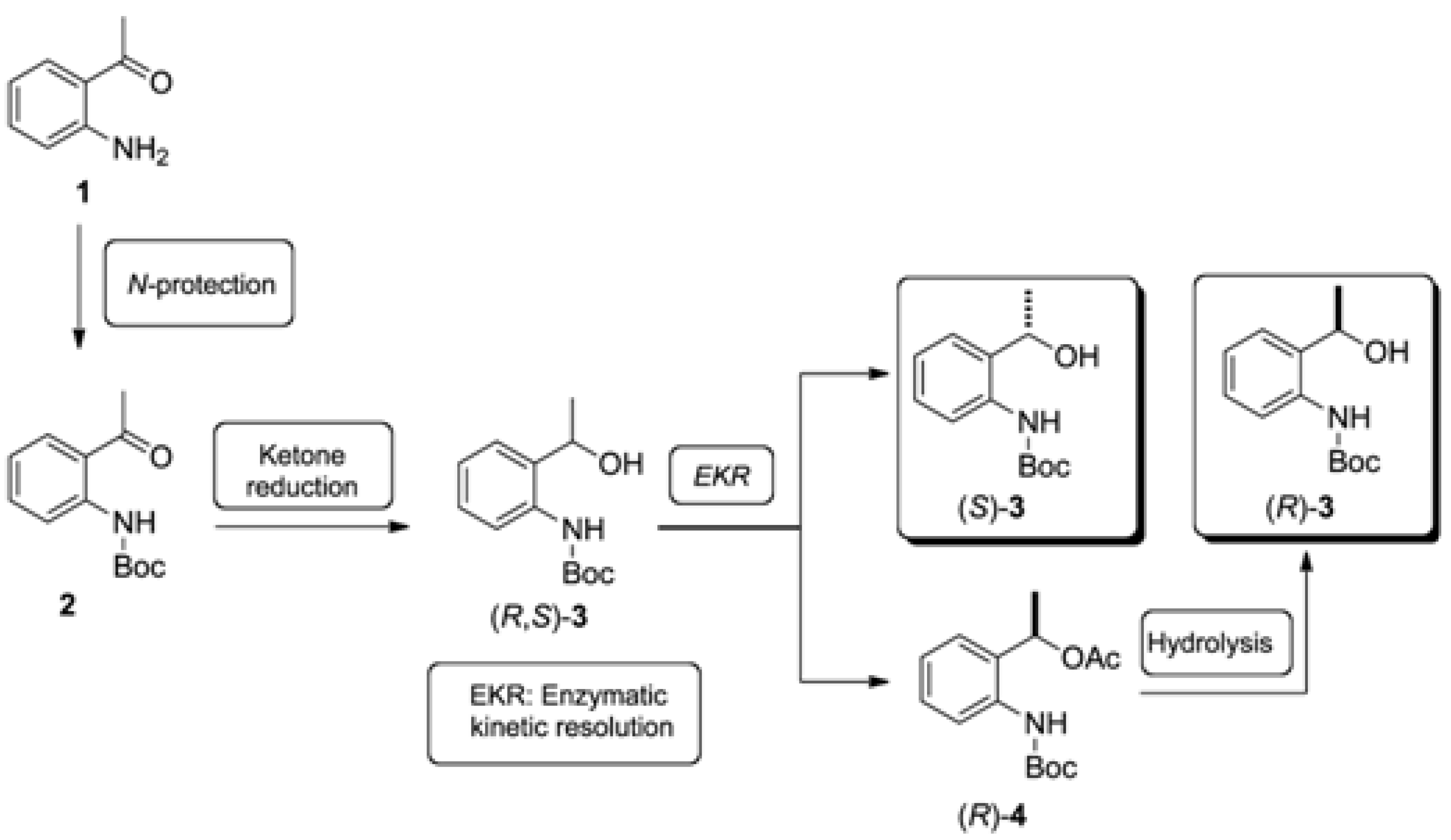
3.2. Synthesis of Racemic tert-butyl (2-(1-Hydroxyethyl)phenyl)carbamate [(R,S)-3]
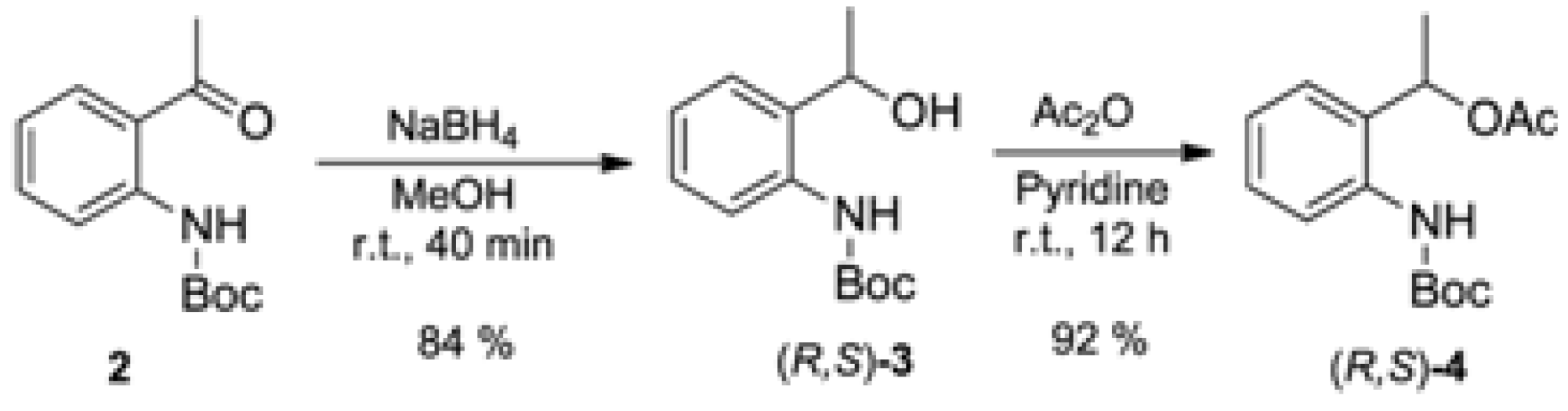
To a solution of tert-butyl (2-acetylphenyl)carbamate (2, 1.175 g, 5 mmol) in methanol (50 mL) NaBH4 (0.21 g, 5.5 mmol) at 0 °C was added. After adding NaBH4, the ice bath was removed and the solution was stirred at room temperature for 2 h then concentrated to dryness. To residue water (30 mL) was added and the pH adjusted to 6.0. In the sequence the mixture was extracted with CH2Cl2 (3 × 15 mL), dried over MgSO4, filtered and concentrated to afford the crude tert-butyl (2-(1-hydroxyethyl)phenyl)carbamate (3). This was purified by flash silica gel column chromatography eluted with hexane/EtOAc 9:1 to afford 3 in 84% yield. 1H-NMR (200 MHz, CDCl3);d (ppm): 8.01 (s, 1H); 7.90 (d, 1H, J = 8.3 Hz); 7.26 (t, 1H, J = 7.5 Hz); 7.14 (d, 1H, J = 7.5 Hz); 7,00 (t, 1H, J = 7.5 Hz); 4,95 (qd, 1H, J = 6.6 Hz); 1.54 (m, 12H). 13C-NMR (50 MHz, CDCl3); d (ppm): 153.5; 136.7; 132.7; 127.9; 126.4; 122.9; 121.4; 80.0; 69.5; 28.2; 22.1. IR (film), cm−1: 3457; 3343; 2979; 1761; 1727; 1524; 1449; 1254. HRMS (ESI), [M+Na]+: Calculated for C13H19NO3Na: 260.1263. Found: 260.1262.
3.3. Synthesis of Racemic 1-(2-((tert-Butoxycarbonyl)amino)phenyl)ethyl acetate [(R,S)-4]
To a solution of the (2-(1-hydroxyethyl)phenyl)carbamate (3, 237 g, 1 mmol) in pyridine (2 mL) was added Ac2O (0.10 g, 1 mmol). The solution was stirred at room temperature overnight then diluted in EtOAc (20 mL) and washed with CuSO4 (5 mL portions) to the complete removal of the pyridine. The organic phase was dried over MgSO4, filtered and concentrated to afford the crude 1-(2-((tert-butoxycarbonyl)amino)phenyl)ethyl acetate (4). The crude material was purified by flash silica gel column chromatography eluted with hexane/EtOAc 9:1 to afford 4 in 92% yield. 1H-NMR (200 MHz, CDCl3); d (ppm): 7.78 (d, 2H, J = 8.1 Hz); 7.66 (s, 1H); 7.32 (m, 2H); 7.12 (td, 1H, JA = 7.5 Hz; JB = 0.8 Hz); 5.98 (qd, 1H, J = 6.4 Hz); 2.0 (s, 3H); 1.61 (d, 3H, J = 6.4 Hz); 1.5 (s, 9H). 13C-NMR (50 MHz, CDCl3); d (ppm): 170.2; 152.8; 135.1; 130.7; 128.1; 126.3; 123.6; 122.9; 79.3; 68.1; 27.6; 20.3; 20.0. IR (film), cm−1: 3432; 3338; 2980; 1731; 1591; 1519; 1453; 1241; 1160. HRMS (ESI), [M+Na]+: Calculated for C15H21NO4Na: 302.1368. Found 302.1364.
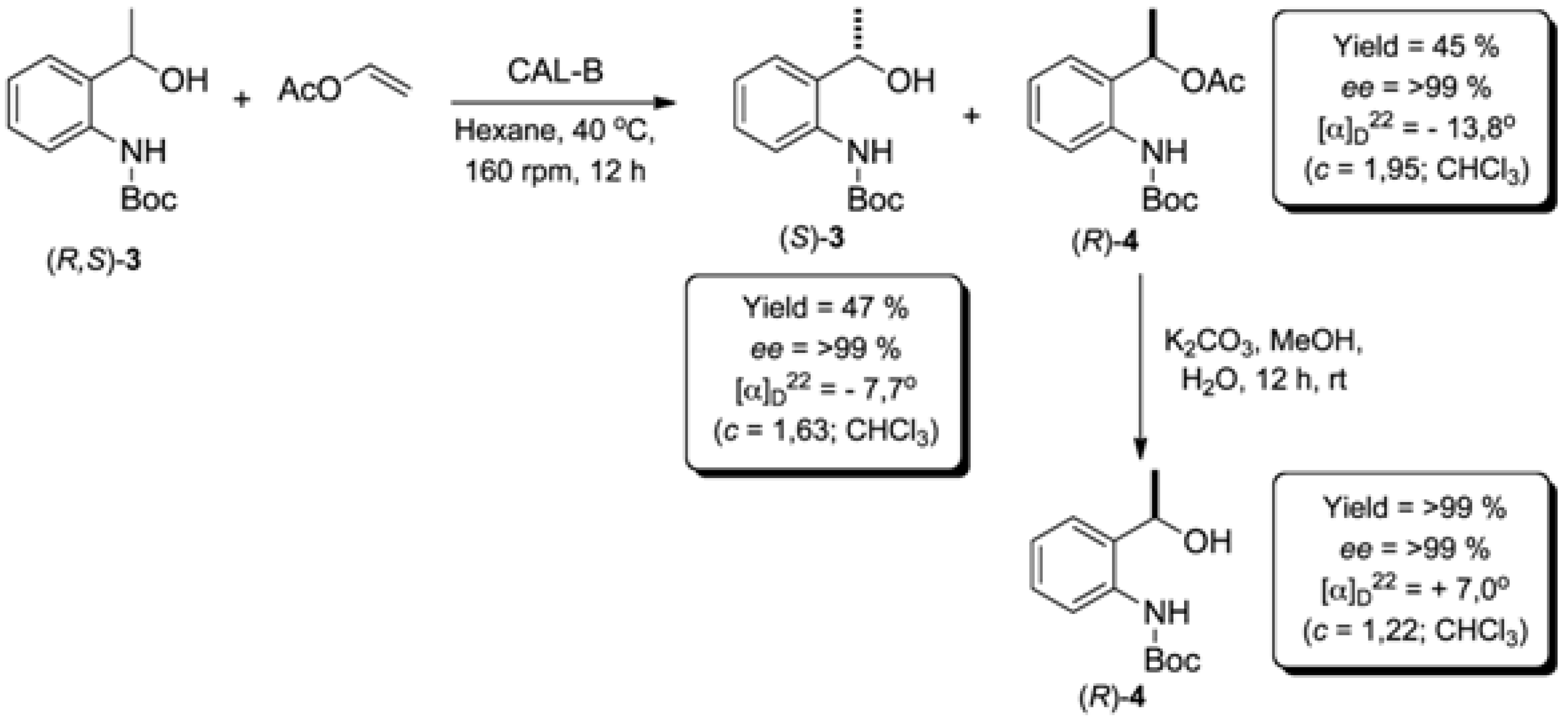
3.4. Enzymatic Kinetic Resolution of the (R,S)-tert-butyl 2-(1-Hydroxyethyl)phenylcarbamate [(R,S)-3]
To solution of racemic tert-butyl (2-(1-hydroxyethyl)phenyl)carbamate (3, 1.185 g; 5 mmol) in hexane (20 mL), CAL-B (Novozym® 435; 400 mg) and vinyl acetate (1.72 g; 20 mmol) were added. The mixture was stirred in an orbital shaker at 40 °C for 12 h (160 rpm). Following that, the enzyme was filtered off and washed with dichloromethane (3 × 20 mL). The solvent was removed under reduced pressure and the residue was purified by flash silica gel column chromatography eluted with hexane/EtOAc 9:1 to afford (S)-3 (ee > 99%) in 47% yield and (R)-4 (ee > 99%) in 45% yield.
3.5. HPLC Analysis of (S)- and (R)-tert-butyl (2-(1-Hydroxyethyl)phenyl)carbamate (3)
HPLC conditions: Chiralcel® OD-H column, n-hexane/i-PrOH (99:1), 1.0 mL min−1, 254 nm UV detector. (S)-3: isolated yield = 45%; retention time: 23.7 min; ee > 99%; [α]D22 = −7.7 (c = 1.63; CHCl3). (R)-3: Isolated yield = 45%; retention time: 29.2 min; ee > 99%; [α]D22 = 7.0 (c = 1.22; CHCl3).
3.6. General Procedure to Remove Boc-Protecting Group (Adapted from Reference [19])
To a mixture of AcOEt:HCl 3 mol L−1 1:1 (5 mL), N-Boc protected compound (1 mmol) was added. The mixture was stirred at room temperature for 1 h. After that, the solvent was removed under vacuum. The residue was dissolved in CH2Cl2 (10 mL) and washed with saturated NaHCO3 solution (3 × 3 mL). Then, the organic layer was dried over MgSO4, filtered and concentrated to dryness under vacuum. The product was obtained in quantitative yield without further purification.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

