Tripolyphosphate Cross-Linked Macromolecular Composites for the Growth of Shape- and Size-Controlled Apatites

Abstract
:1. Introduction
2. Results and Discussion
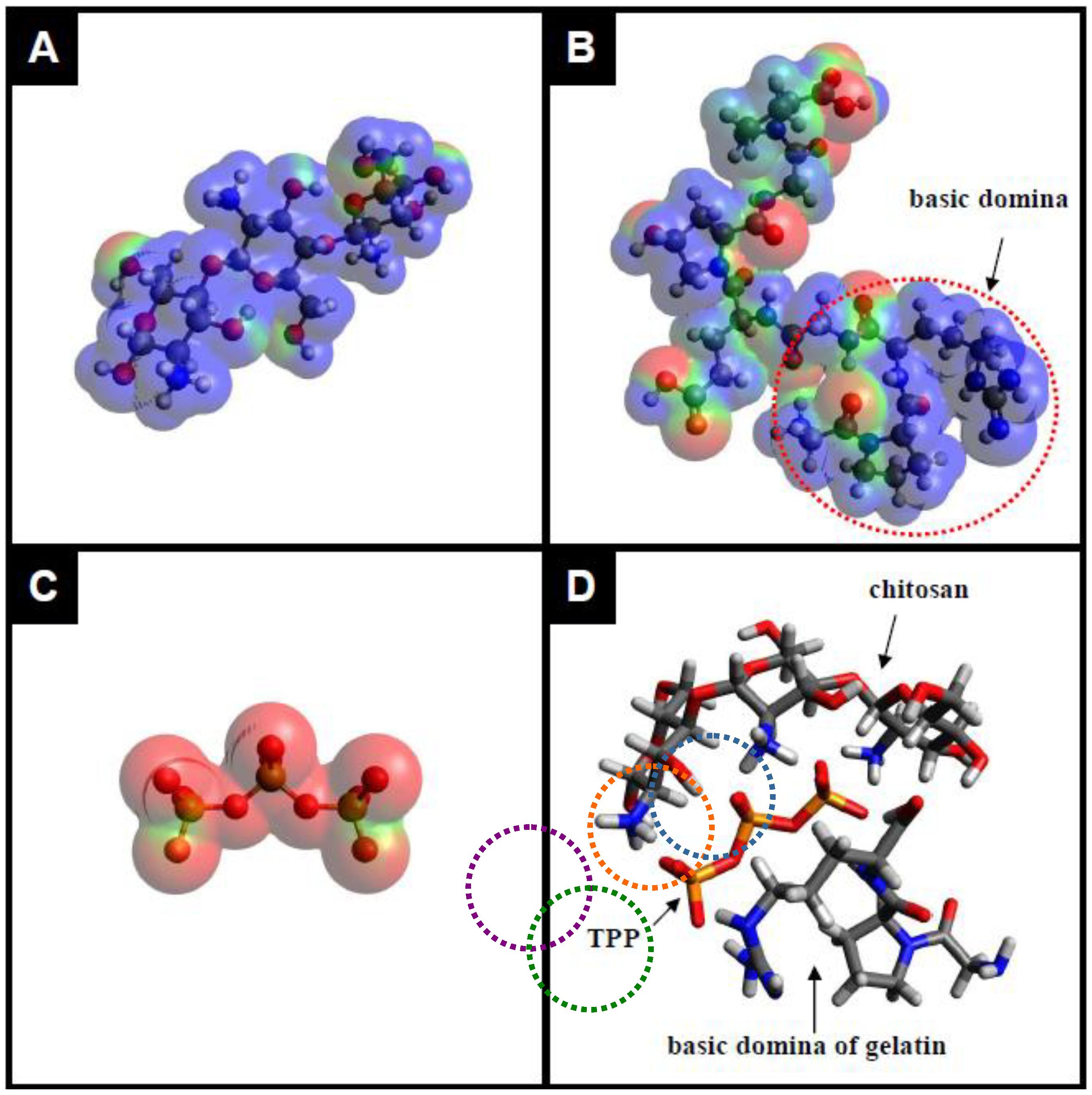
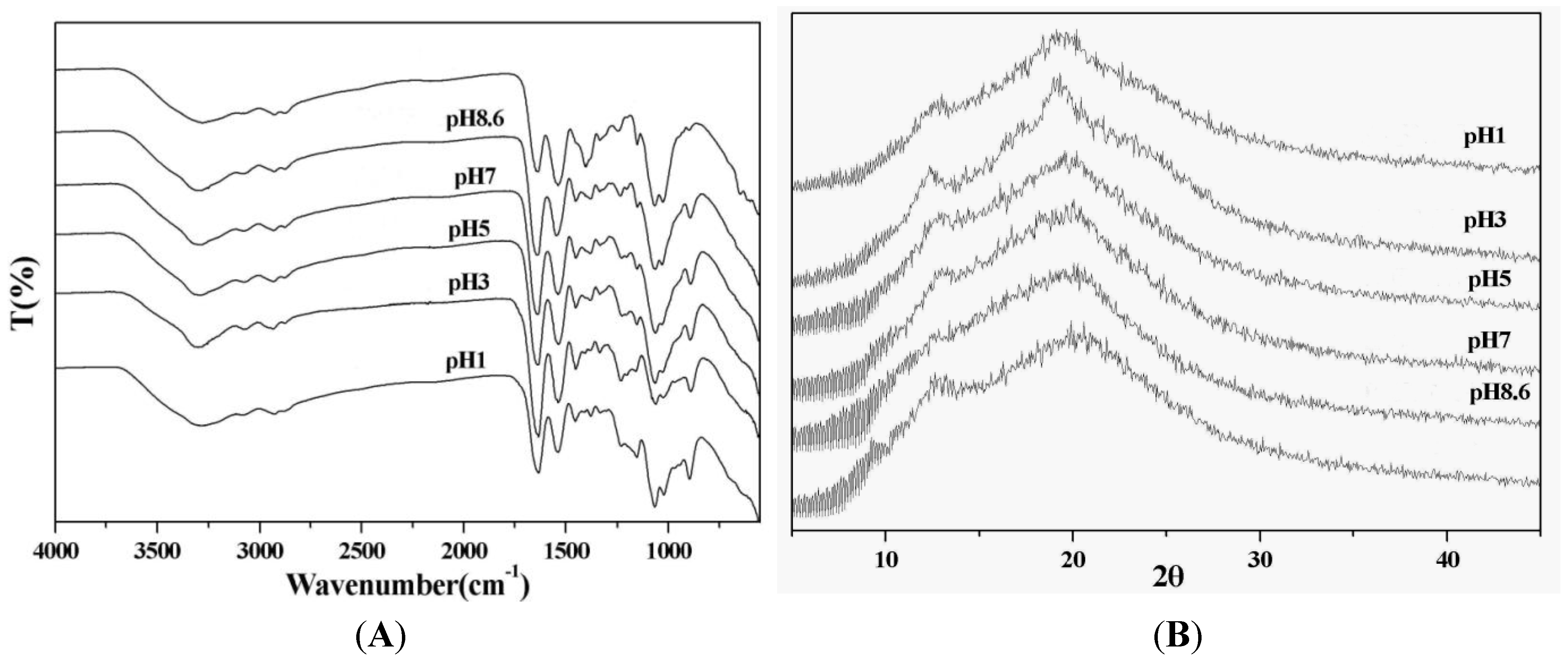
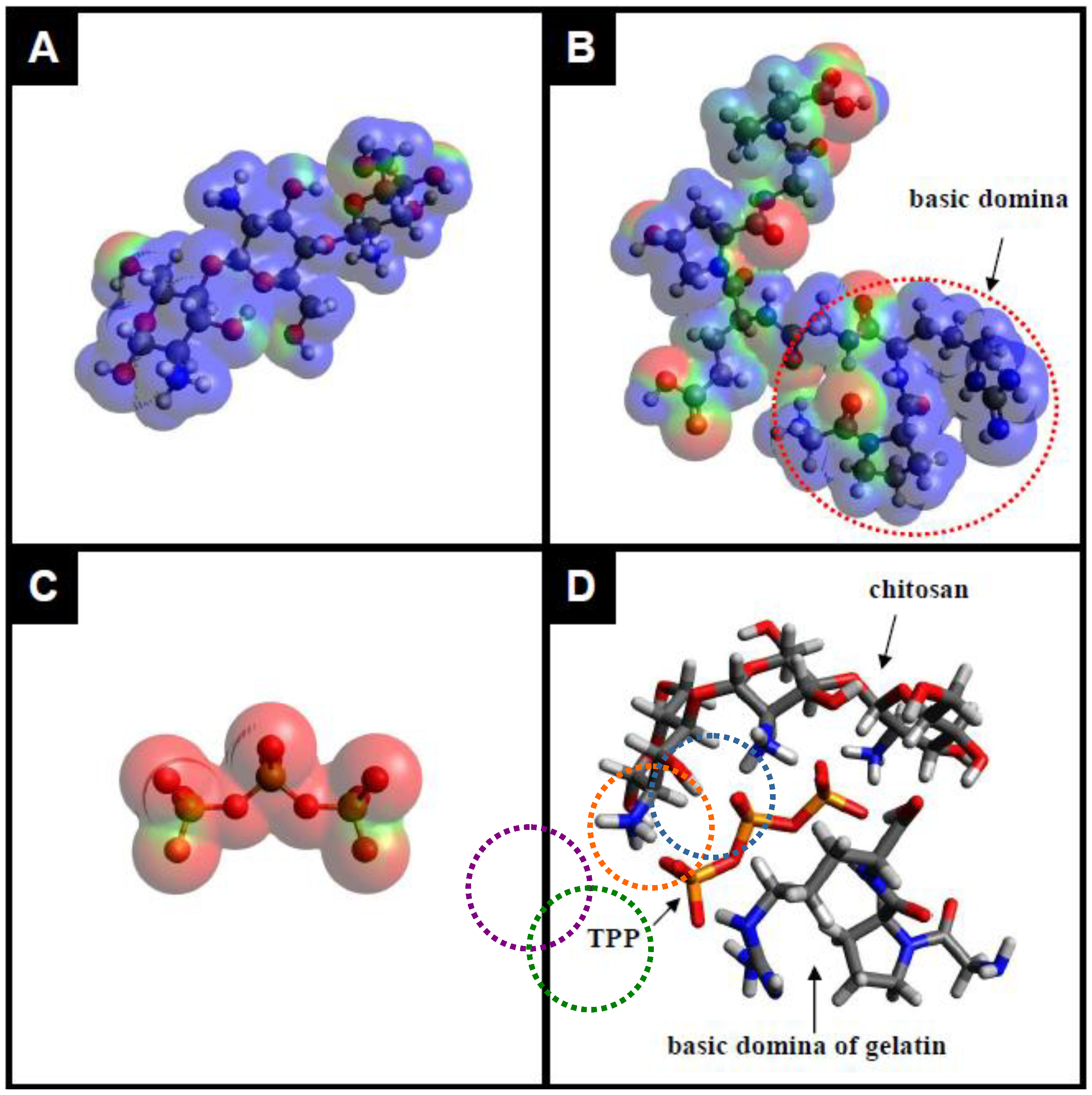
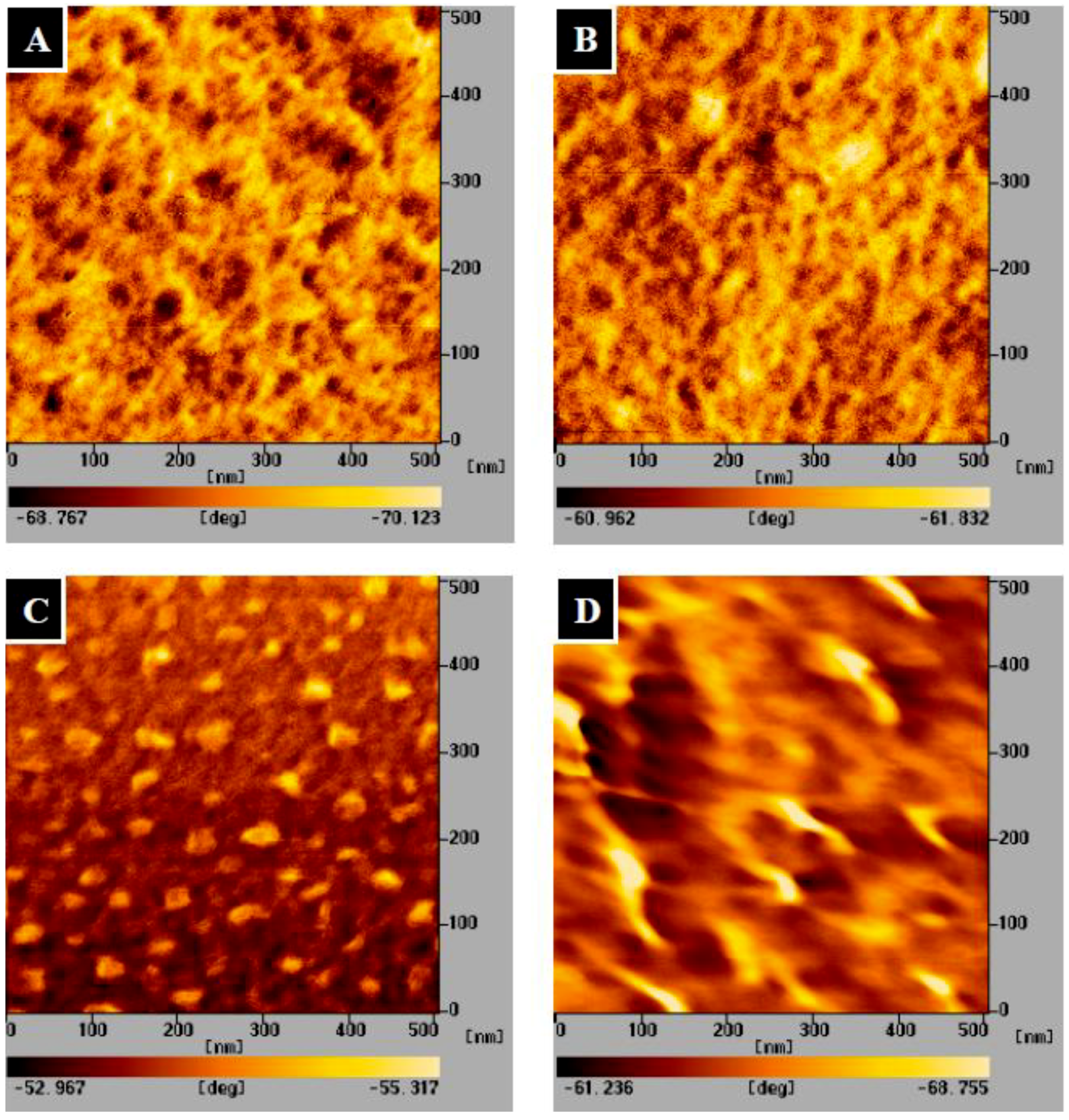
2.1. Preparation and Characterization of the TPP-CG Composites
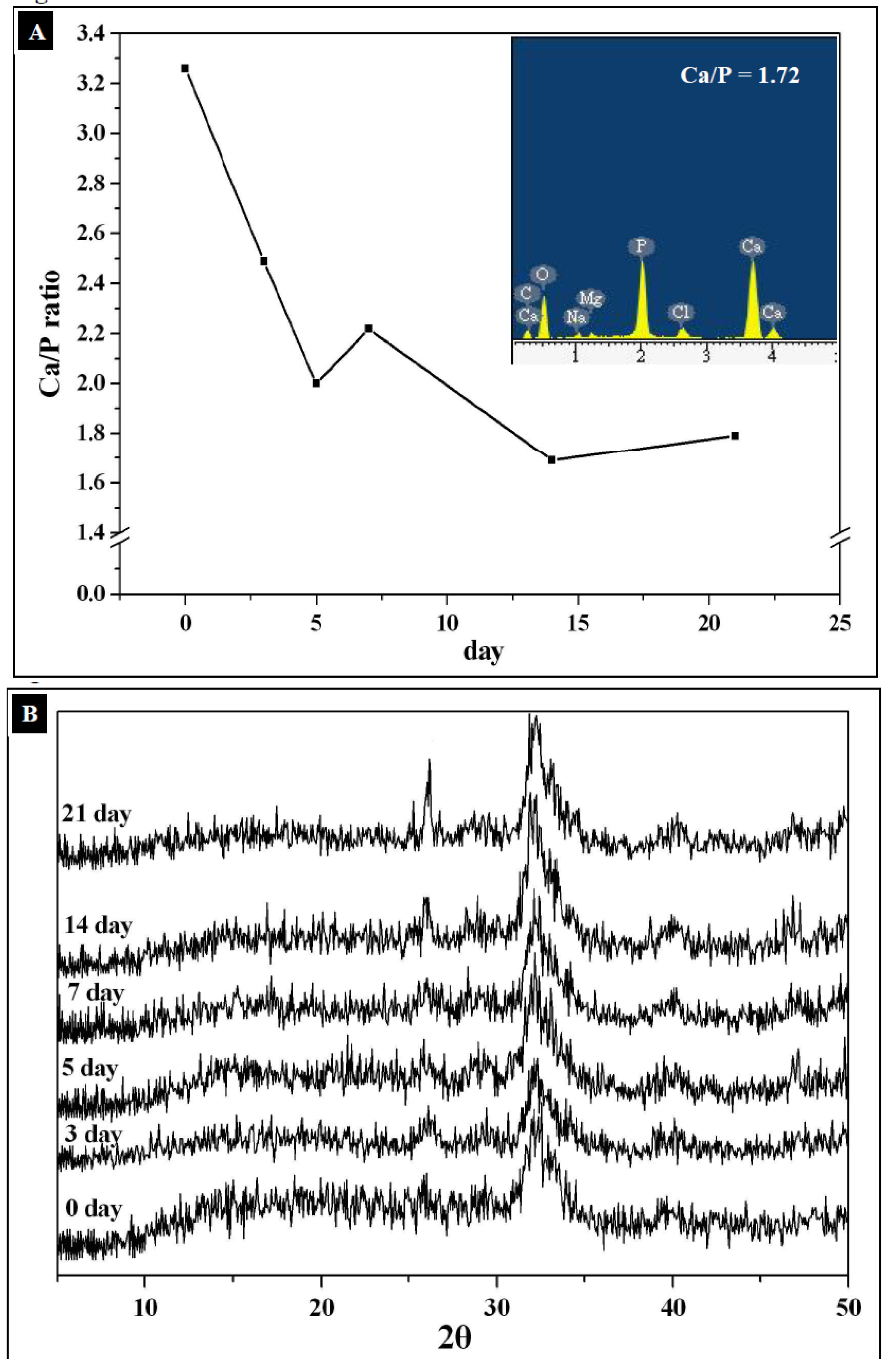
2.2. Characterization of Mineralized Calcium Phosphates
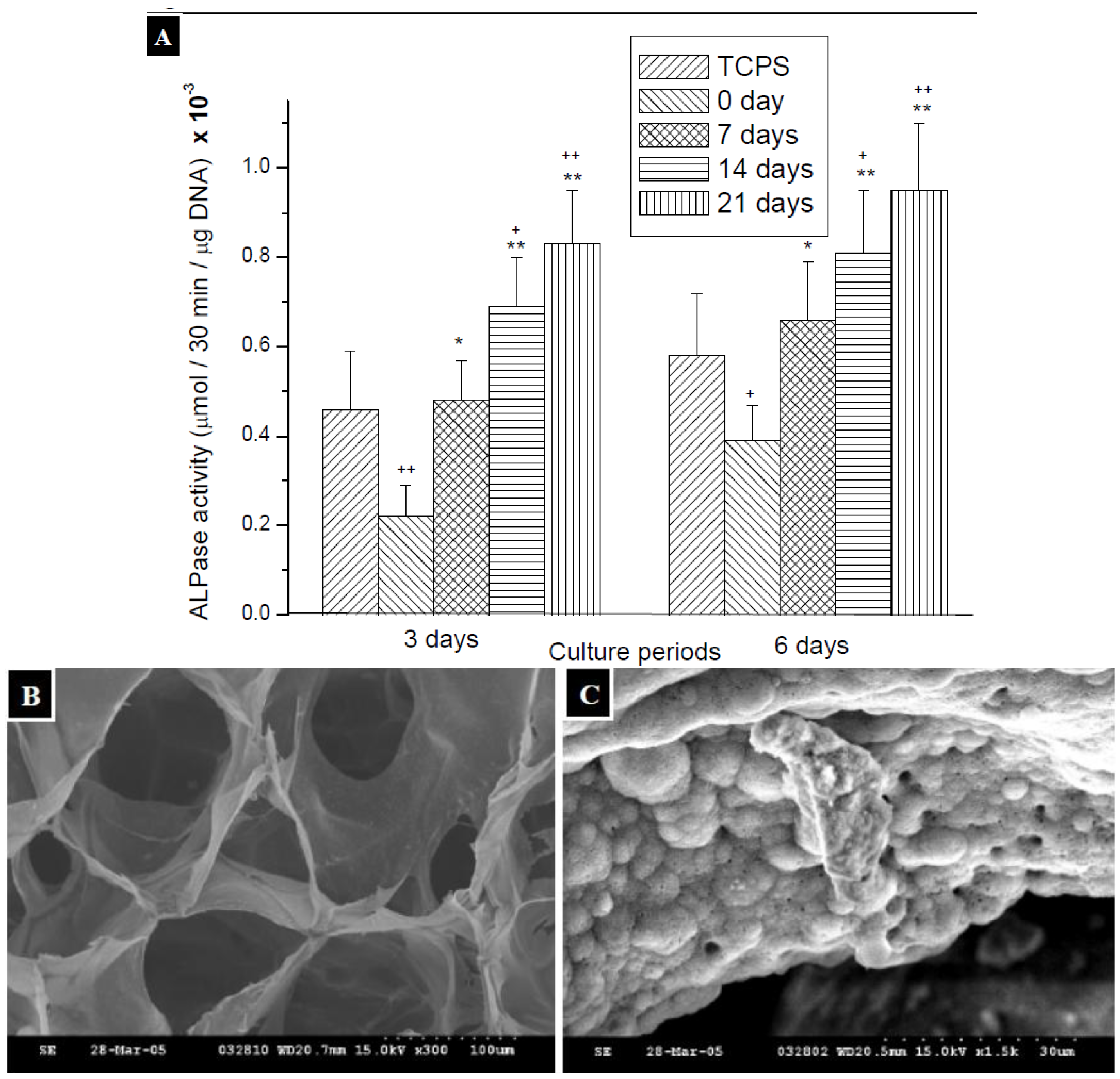
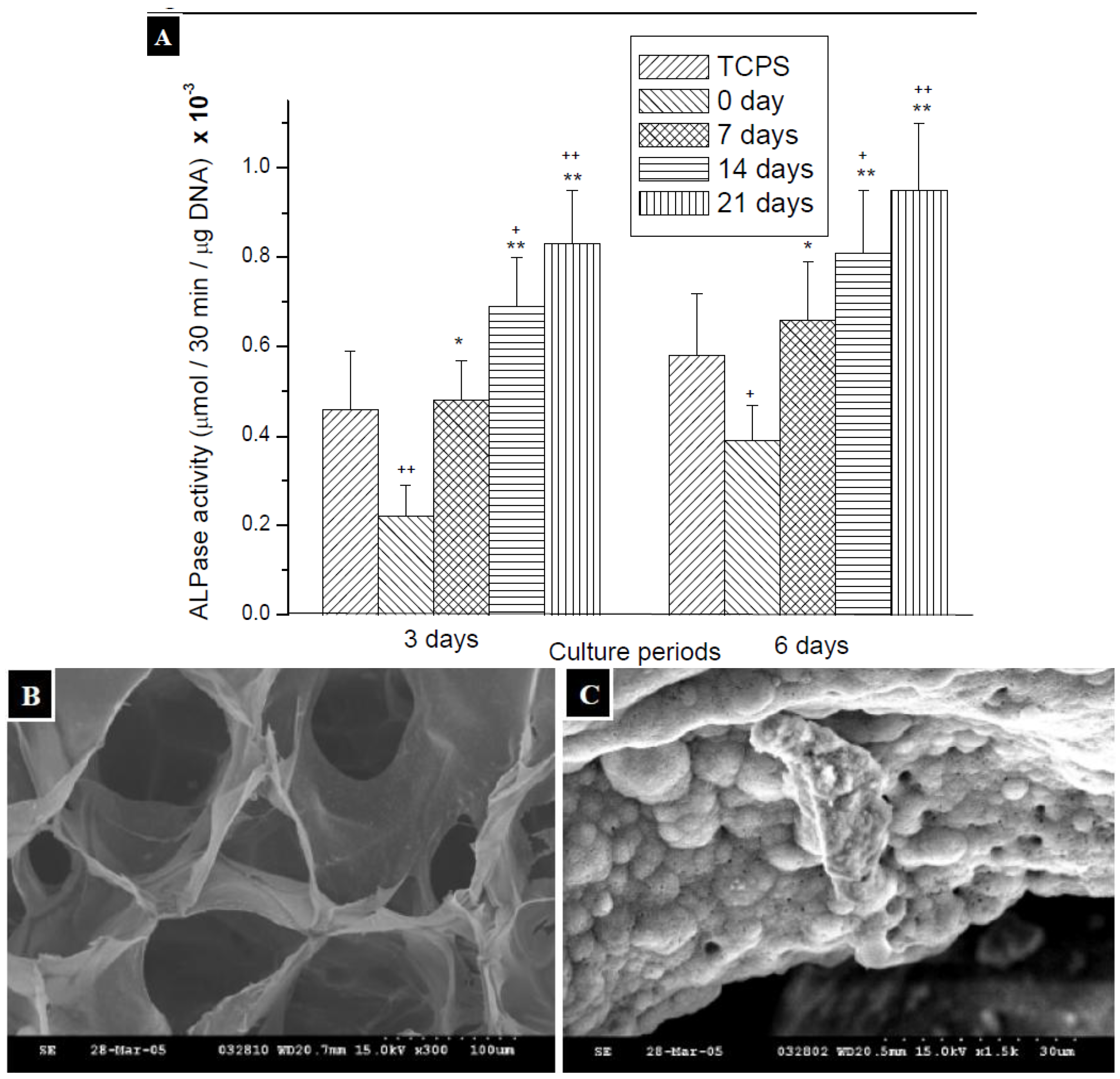
2.3. Measurement of ALPase Activity
3. Experimental
3.1. Materials
3.2. Preparation of TPP-CG Composites
3.3. Characterization of the TPP-CG Composites
3.4. Biomineralization
3.5. Characterization of Mineralized Calcium Phosphates on/in the Biotemplate
3.6. Determination of Crystallites Size of Hydroxyapatite (HAp)
3.7. Measurement of ALPase Activity
4. Conclusions
Acknowledgments
References
- Capito, R.M.; Azevedo, H.S.; Velichko, Y.S.; Mata, A.; Stupp, S.I. Self-assembly of Large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. [Google Scholar] [CrossRef] [PubMed]
- Safinya, C.R. Structures of lipid-DNA complexes: Supramolecular assembly and gene delivery. Curr. Opin. Struct. Biol. 2001, 11, 440–448. [Google Scholar] [CrossRef]
- Lee, K.Y.; Kong, H.J.; Larson, R.G.; Mooney, D.J. Hydrogel formation via cell cross-linking. Adv. Mater. 2003, 15, 1828–1832. [Google Scholar] [CrossRef]
- Sun, W.; Puzas, J.E.; Sheu, T.J.; Liu, X.; Fauchet, P.M. Nano- and microscale porous silicon as a cell interface for bone-tissue engineering. Adv. Mater. 2007, 19, 921–924. [Google Scholar] [CrossRef]
- Busch, S.; Schwarz, U.; Kniep, R. Chemical and structural investigations of biomimetically grown fluorapatite-gelatin-composite aggregates. Adv. Funct. Mater. 2003, 13, 189–198. [Google Scholar] [CrossRef]
- Stupp, S.I.; Braun, P.V. Molecular manipulation of microstructures: Biomaterials, ceramics, and semiconductors. Science 1997, 277, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.W.; Antonietti, M.; Yu, S.H.; Cölfen, H. Polymer-mediated mineralzation and self-similar mesoscale-organized calcium carbonate with unusual superstructures. Adv. Mater. 2008, 20, 1333–1338. [Google Scholar] [CrossRef]
- Hu, Q.; Li, B.; Wang, M.; Shen, J. Prepration and characterization of biodegradable chitosan/hydroxyapatite nanocomposite rods via in situ hybridization: A potential material as internal fixation of bone fracture. Biomaterials 2004, 25, 779–785. [Google Scholar] [CrossRef]
- Kikuchi, M.; Matsumoto, H.N.; Yamada, T.; Koyama, Y.; Takakuda, K.; Tanaka, J. Glutaraldehyde cross-linked hydroxyapatite/collagen self-organized nano-composites. Biomaterials 2004, 25, 63–69. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, X.; Fu, R.K.Y.; Ho, J.P.Y.; Ding, C.; Chu, P.K. Plasma-treated nanostructured TiO2 surface supporting biomimetic growth of apatite. Biomaterials 2005, 26, 6143–6150. [Google Scholar] [CrossRef] [PubMed]
- Balani, K.; Chen, Y.; Harimkar, S.P.; Dahotre, N.B.; Agarwal, A. Tribological behavior of plasma-sprayed carbon nanotube-reinforced hydroxyapatite coating in physiological solution. Acta Biomater. 2007, 3, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.K.; Hauschka, P.V.; Poole, A.R.; Rosenberg, L.C.; Goldberg, H.A. Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem. J. 1996, 317, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Segman-Magidovich, S.; Grisaru, H.; Gitli, T.; Levi-Kalisman, Y.; Rapaport, H. Matrices of acidic beta-sheet peptides as templates for calcium phosphate mineralization. Adv. Mater. 2008, 20, 2156–2161. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Cheng, W.; Yang, Y.; Zhu, M.; Zhou, C. Novel injectable calcium phosphate/chitosan composites for bone substitute materials. Acta Biomater. 2006, 2, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Tong, H.; Shen, X.; Chen, W.; Yan, J.; Hu, J. Preparation and characterization of homogeneous chitosan-polylactic acid/hydroxyapatite nanocomposite for bone tissue engineering and evaluation of its mechanical properties. Acta Biomater. 2009, 5, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Rusu, V.M.; Ng, C.H.; Wilke, M.; Tiersch, B.; Fratzl, P.; Peter, M.G. Size-controlled hydroxyapatite nanoparticles as self-organized organic-inorganic composite materials. Biomaterials 2005, 26, 5414–5426. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Gao, Y.; Lu, G.; Gong, Y.; Zhao, N.; Zhang, X. A study on the bioactivity of chitosan/nano-hydroxyapatite composite scaffolds for bone tissue engineering. Eur. Polym. J. 2006, 42, 3171–3179. [Google Scholar] [CrossRef]
- Cai, Y.; Tang, R. Calcium phosphate nanoparticles in biomineralization and biomaterials. J. Mater. Chem. 2008, 18, 3775–3787. [Google Scholar] [CrossRef]
- Hu, Q.; Tan, Z.; Liu, Y.; Tao, J.; Cai, Y.; Zhang, M.; Pan, H.; Xu, X.; Tang, R. Effect of crystallinity of calcium phosphate nanoparticles on adhesion, proliferation, and differentiation of bone marrow mesenchymal stem cells. J. Mater. Chem. 2007, 17, 4690–4698. [Google Scholar] [CrossRef]
- Scharnweber, T.; Santos, C.; Franke, R.P.; Almeida, M.M.; Costa, M.E. Influence of spray-dried hydroxyapatite-5-fluorouracil granules on cell lines derived from tissues of mesenchymal origin. Molecules 2008, 13, 2729–2739. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, S.E.; Choi, H.W.; Kim, C.W.; Kim, K.J.; Lee, S.C. The effect of surface-modified nano-hydroxyapatite on biocompatibility of poly(ε-caprolactone)/hydroxyapatite nanocomposites. Eur. Polym. J. 2007, 43, 1602–1608. [Google Scholar] [CrossRef]
- Kalfus, J.; Jancar, J. Immobilization of polyvinylacetate macromolecules on hydroxyapatite nanoparticles. Polymer 2007, 48, 3935–3937. [Google Scholar] [CrossRef]
- Qiu, X.; Hong, Z.; Hu, J.; Chen, L.; Chen, X.; Jing, X. Hydroxyapatite surface modified by L-lactic acid and its subsequent grafting polymerization of L-lactide. Biomacromolecules 2005, 6, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.; Ramakrishna, S. Bioresorbable composite bone paste using polysaccharide based nano hydroxyapatite. Biomaterials 2004, 25, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.L. Synthesis and characterization of a novel chitosan-gelatin bioconjugate with fluorescence emission. Biomacromolecules 2005, 6, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.L.; Wu, Y.Y.; Lin, Y.H.; Sonaje, K.; Ho, Y.C.; Chen, C.T.; Juang, J.H.; Sung, H.W. Oral delivery of peptide drugs using nanoparticles self-assembled by poly(gamma-glutamic acid) and a chitosan derivative functionalized by trimethylation. Bioconjug. Chem. 2008, 19, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.W.; Yu, S.H.; Ho, Y.C.; Mi, F.L.; Kuo, P.L.; Sung, H.W. Heparinized chitosan/poly(gamma-glutamic acid) nanoparticles for multi-functional delivery of fibroblast growth factor and heparin. Biomaterials 2010, 31, 9320–9332. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Liao, Z.X.; Panda, N.; Tang, D.W.; Yu, S.H.; Mi, F.L.; Sung, H.W. Self-organized nanoparticles prepared by guanidine- and disulfide-modified chitosan as a gene delivery carrier. J. Mater. Chem. 2011, 21, 16918–16927. [Google Scholar] [CrossRef]
- Yu, S.H.; Mi, F.L.; Pang, J.C.; Jiang, S.C.; Kuo, T.H.; Wu, S.J.; Shyu, S.S. Preparation and characterization of radical and pH-responsive chitosan-gallic acid conjugate drug carriers. Carbohydr. Polym. 2011, 84, 794–802. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Glimcher, M.J.; Cooper, R.R.; Recker, R. Bone biology. I: Structure, blood supply, cells, matrix, and mineralization. Instr. Course Lect. 1996, 45, 371–386. [Google Scholar] [PubMed]
- Du, C.; Cui, F.Z.; Zhang, W.; Feng, Q.L.; Zhu, X.D.; de Groot, K. Formation of calcium phosphate/collagen composites through mineralization of collagen matrix. J. Biomed. Mater. Res. 2000, 50, 518–527. [Google Scholar] [CrossRef]
- Isama, K.; Tsuchiya, T. Enhancing effect of poly(L-lactide) on the differentiation of mouse osteoblast-like MC3T3-E1 cells. Biomaterials 2003, 24, 3303–3309. [Google Scholar] [CrossRef]
- Hou, L.T.; Liu, C.M.; Lei, J.Y.; Wong, M.Y.; Chen, J.K. Biological effects of cementum and bone extracts on human periodontal fibroblasts. J. Periodontol. 2000, 71, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (day) | Line width (002) FWHM (°) | Cristallinity (Xc) | Line width (002) FWHM (rad) | Average crystallite size, L (nm) by Scherrer’s equation |
|---|---|---|---|---|
| 0 | - | - | - | - |
| 3 | 0.98536 | 0.0144 | 0.0172 | 9.2 |
| 5 | 0.86683 | 0.0212 | 0.0151 | 10.5 |
| 7 | 0.76252 | 0.0312 | 0.0133 | 11.9 |
| 14 | 0.52156 | 0.0974 | 0.0091 | 17.4 |
| 21 | 0.44592 | 0.1559 | 0.0078 | 20.3 |
© 2013 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, S.-H.; Wu, S.-J.; Wu, J.-Y.; Peng, C.-K.; Mi, F.-L. Tripolyphosphate Cross-Linked Macromolecular Composites for the Growth of Shape- and Size-Controlled Apatites. Molecules 2013, 18, 27-40. https://doi.org/10.3390/molecules18010027
Yu S-H, Wu S-J, Wu J-Y, Peng C-K, Mi F-L. Tripolyphosphate Cross-Linked Macromolecular Composites for the Growth of Shape- and Size-Controlled Apatites. Molecules. 2013; 18(1):27-40. https://doi.org/10.3390/molecules18010027
Chicago/Turabian StyleYu, Shu-Huei, Shao-Jung Wu, Jui-Yu Wu, Chih-Kang Peng, and Fwu-Long Mi. 2013. "Tripolyphosphate Cross-Linked Macromolecular Composites for the Growth of Shape- and Size-Controlled Apatites" Molecules 18, no. 1: 27-40. https://doi.org/10.3390/molecules18010027
APA StyleYu, S. -H., Wu, S. -J., Wu, J. -Y., Peng, C. -K., & Mi, F. -L. (2013). Tripolyphosphate Cross-Linked Macromolecular Composites for the Growth of Shape- and Size-Controlled Apatites. Molecules, 18(1), 27-40. https://doi.org/10.3390/molecules18010027