
Probing Steroidal Substrate Specificity of Cytochrome P450 BM3 Variants


Abstract
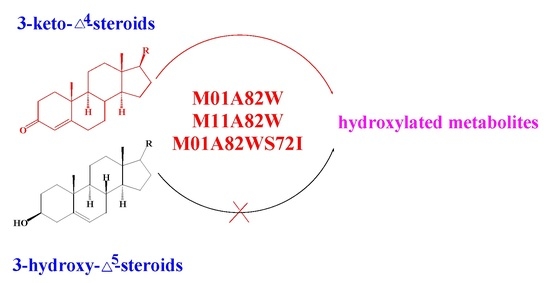
:
1. Introduction
2. Results and Discussion
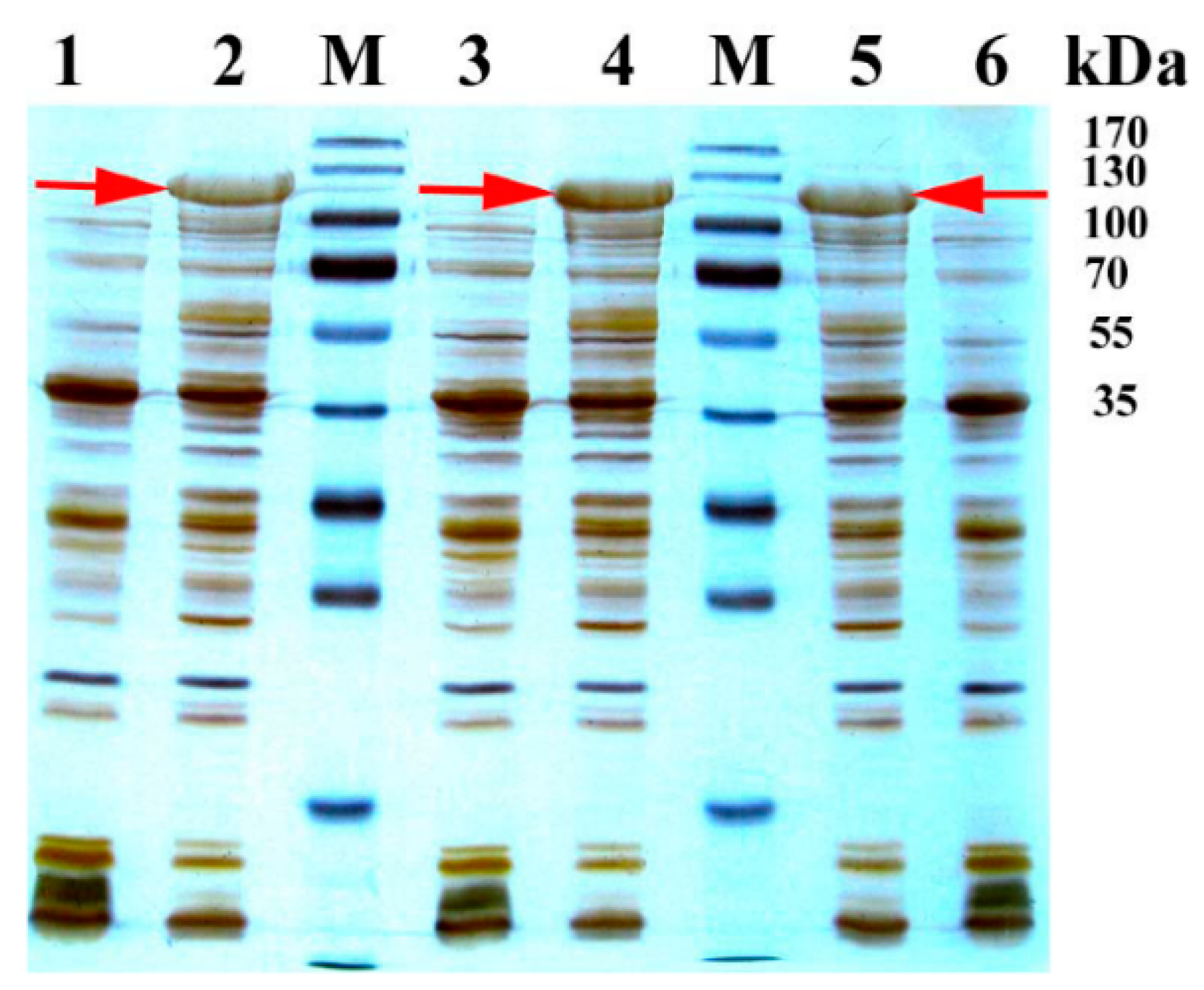
2.1. Expression and Purification of BM3 Mutants
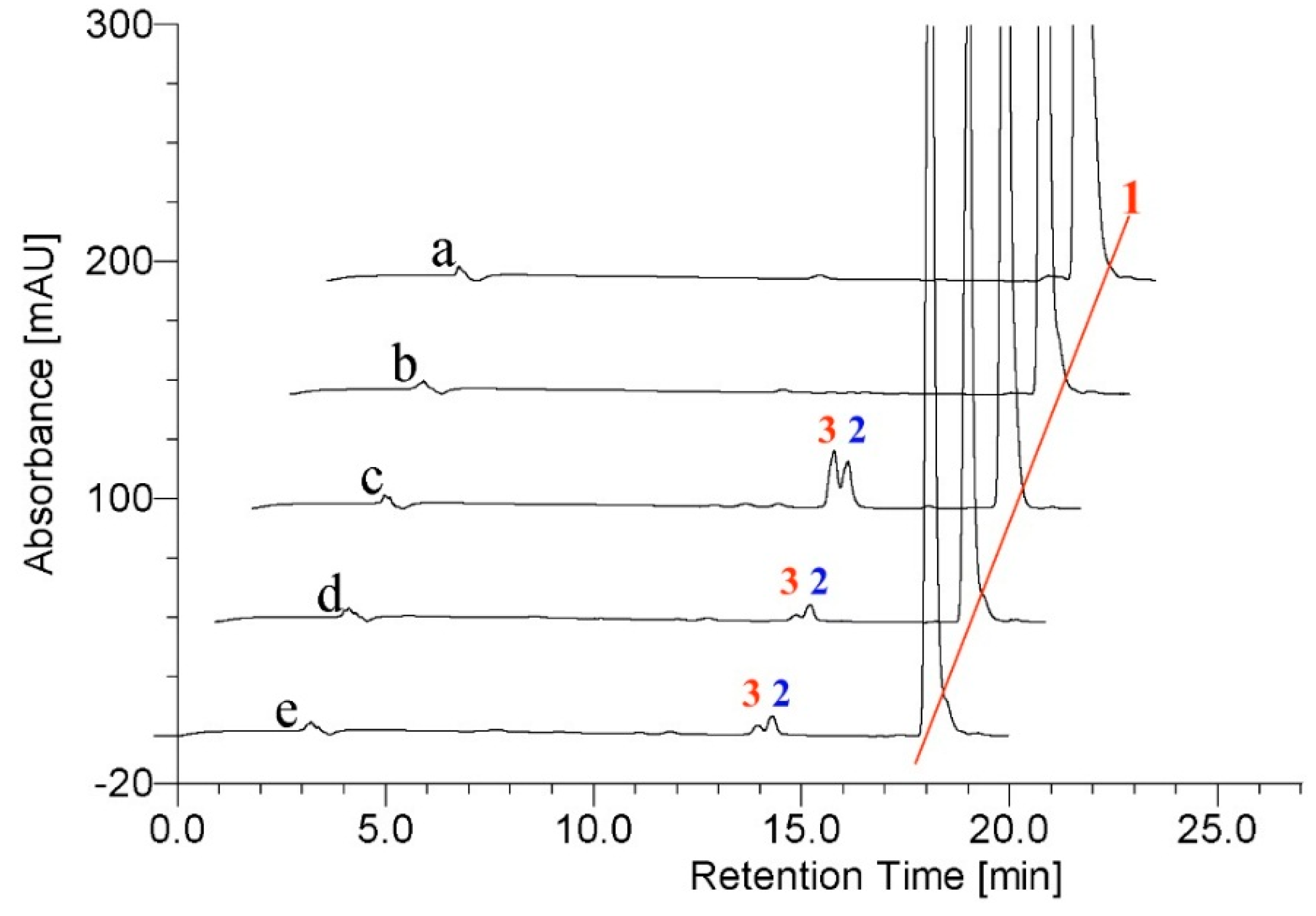
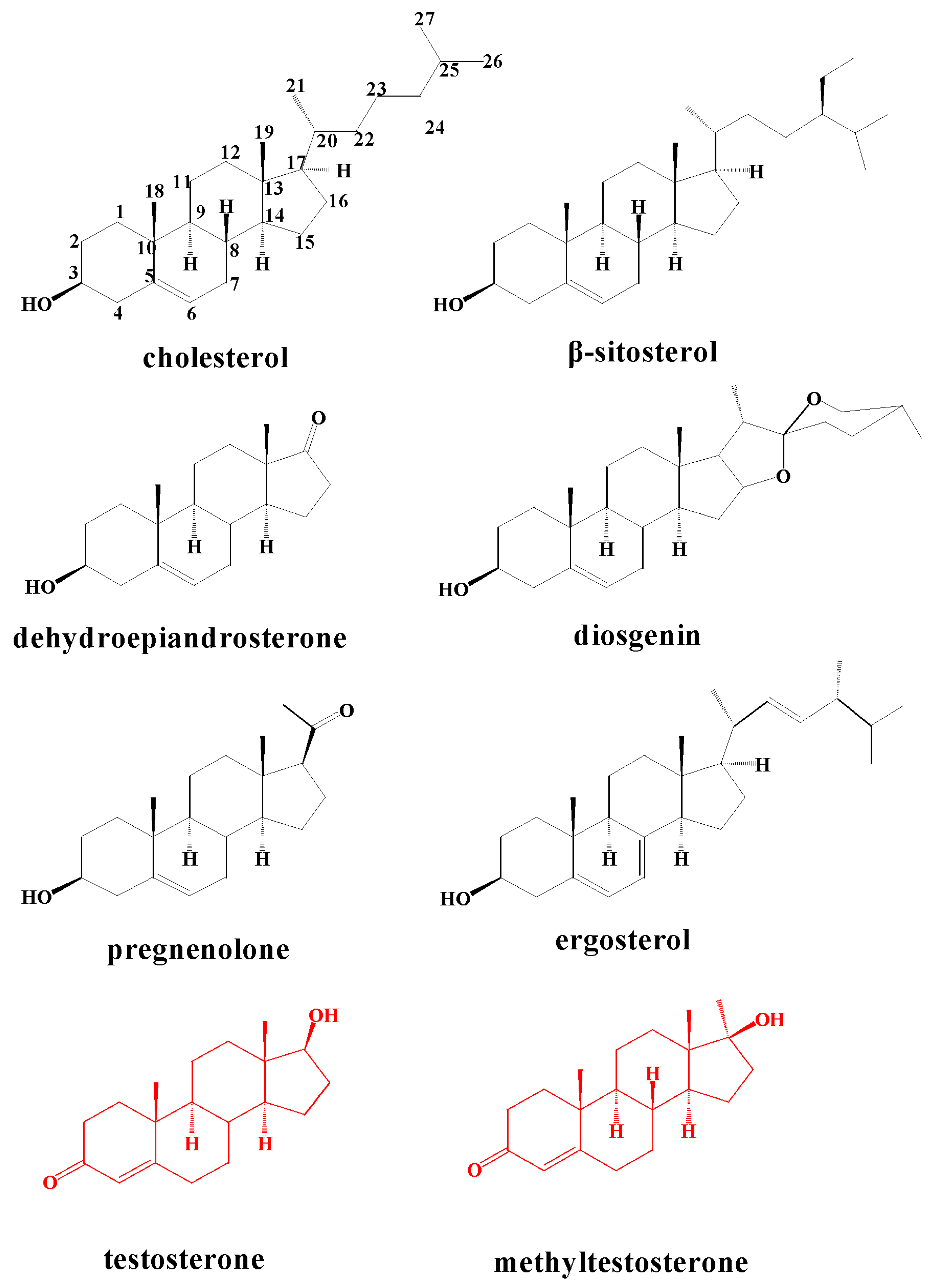
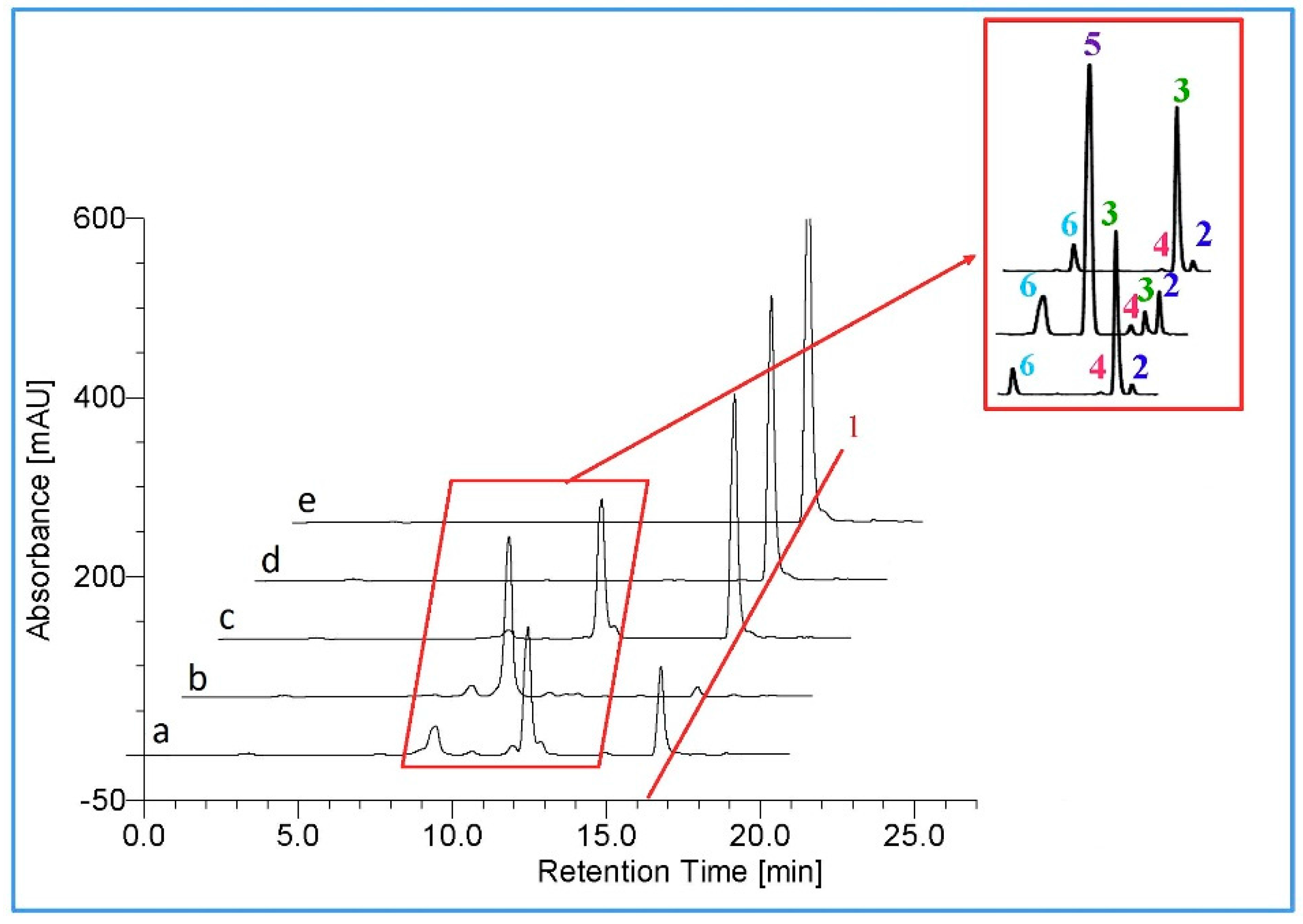
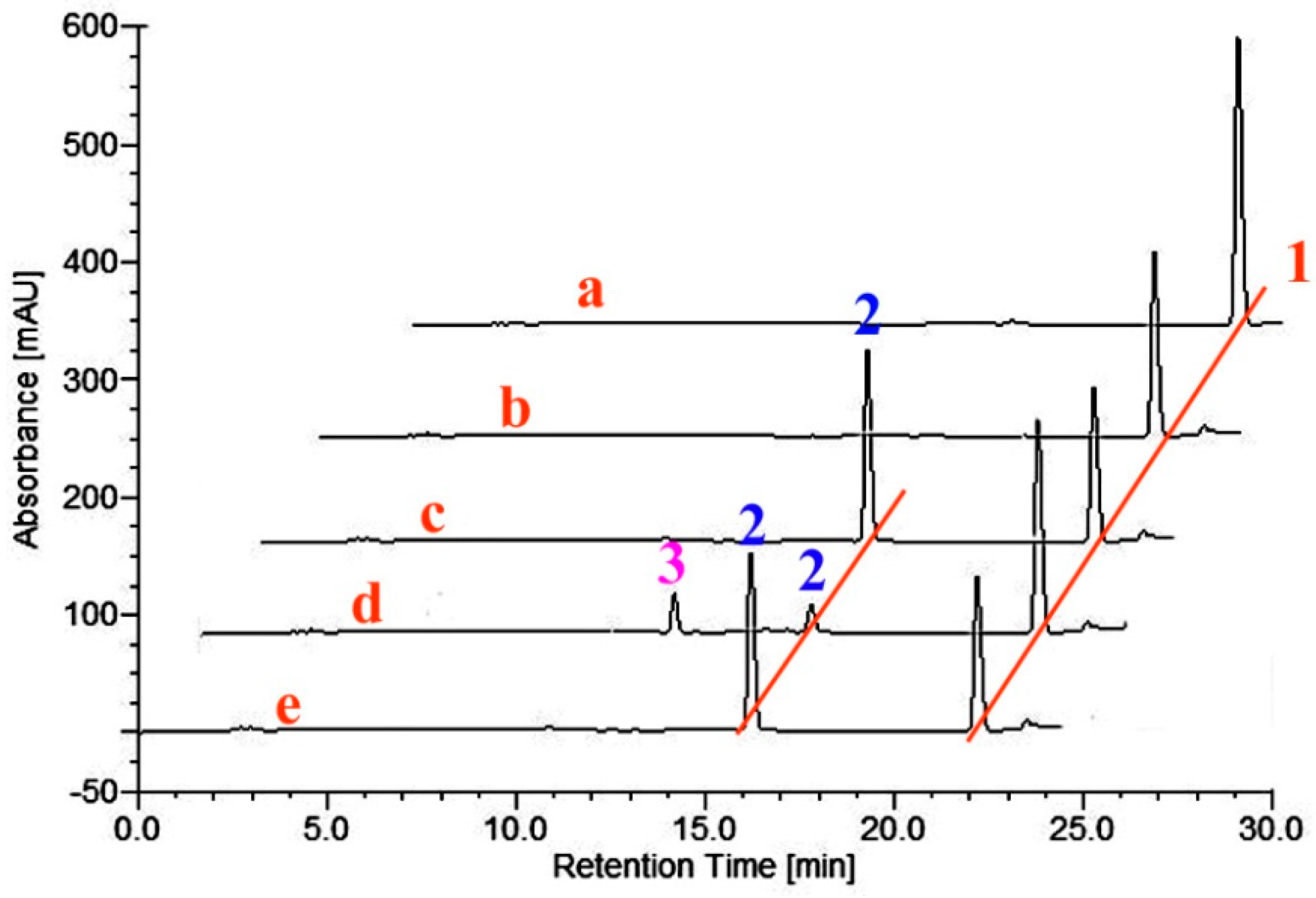
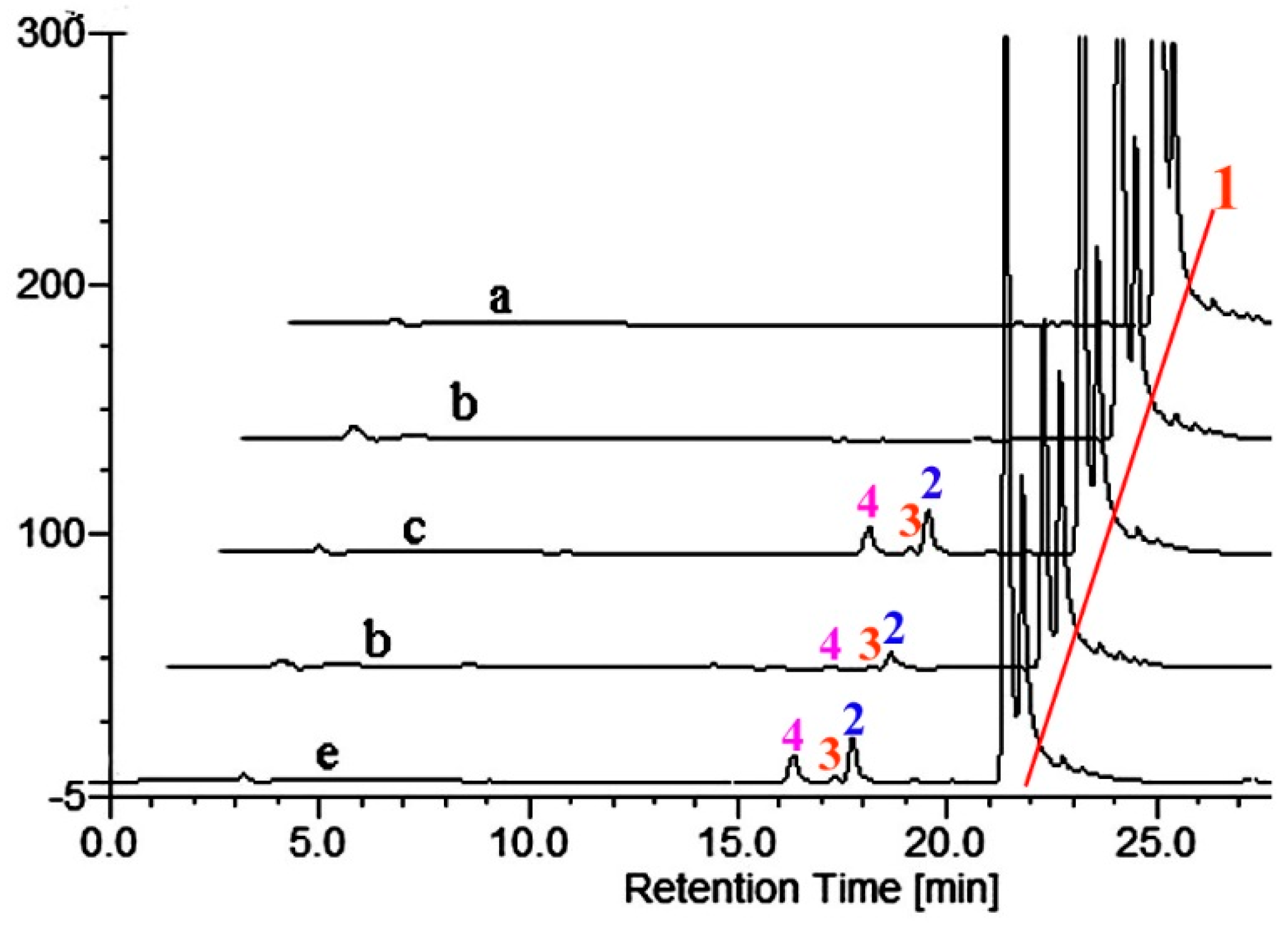
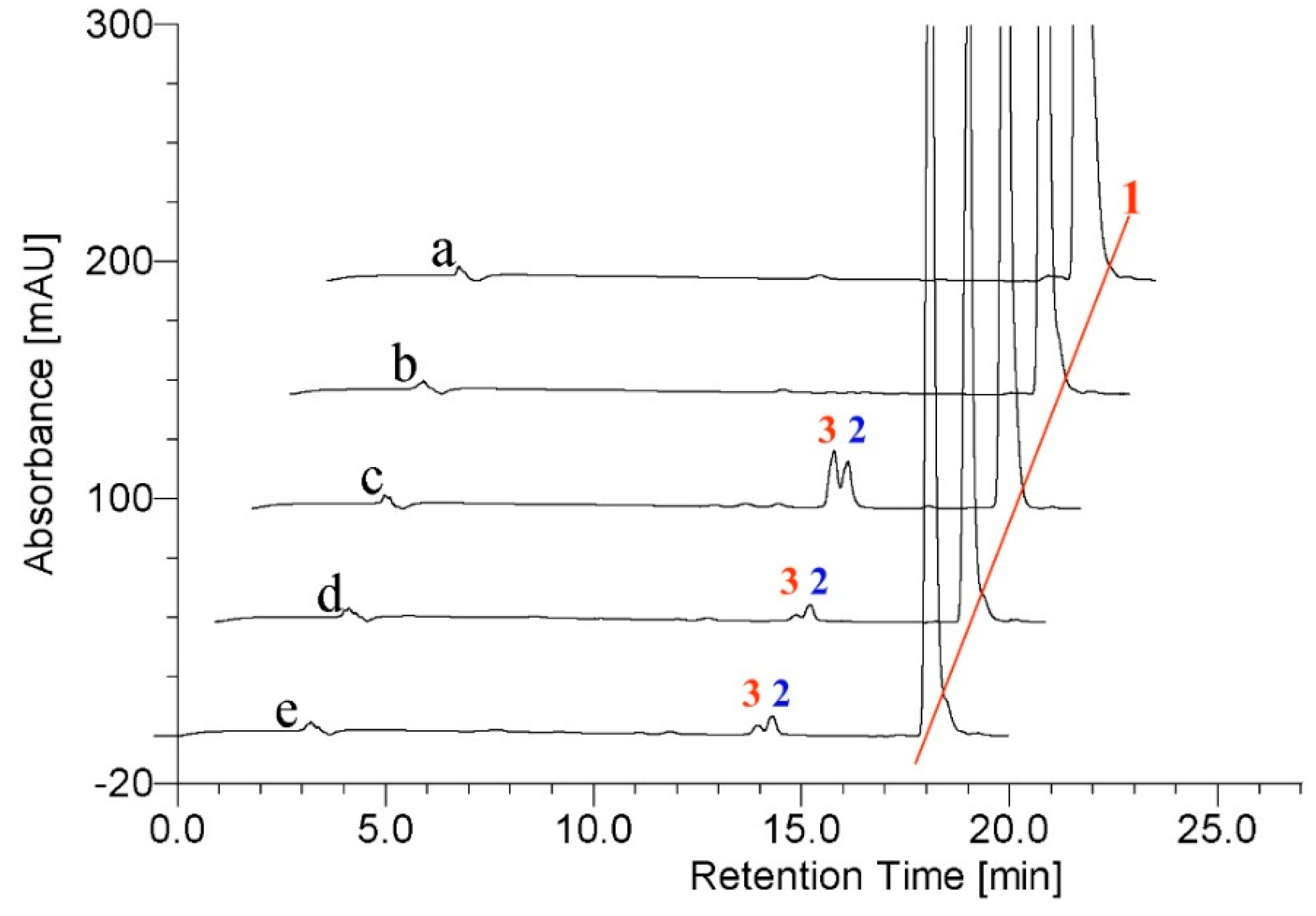
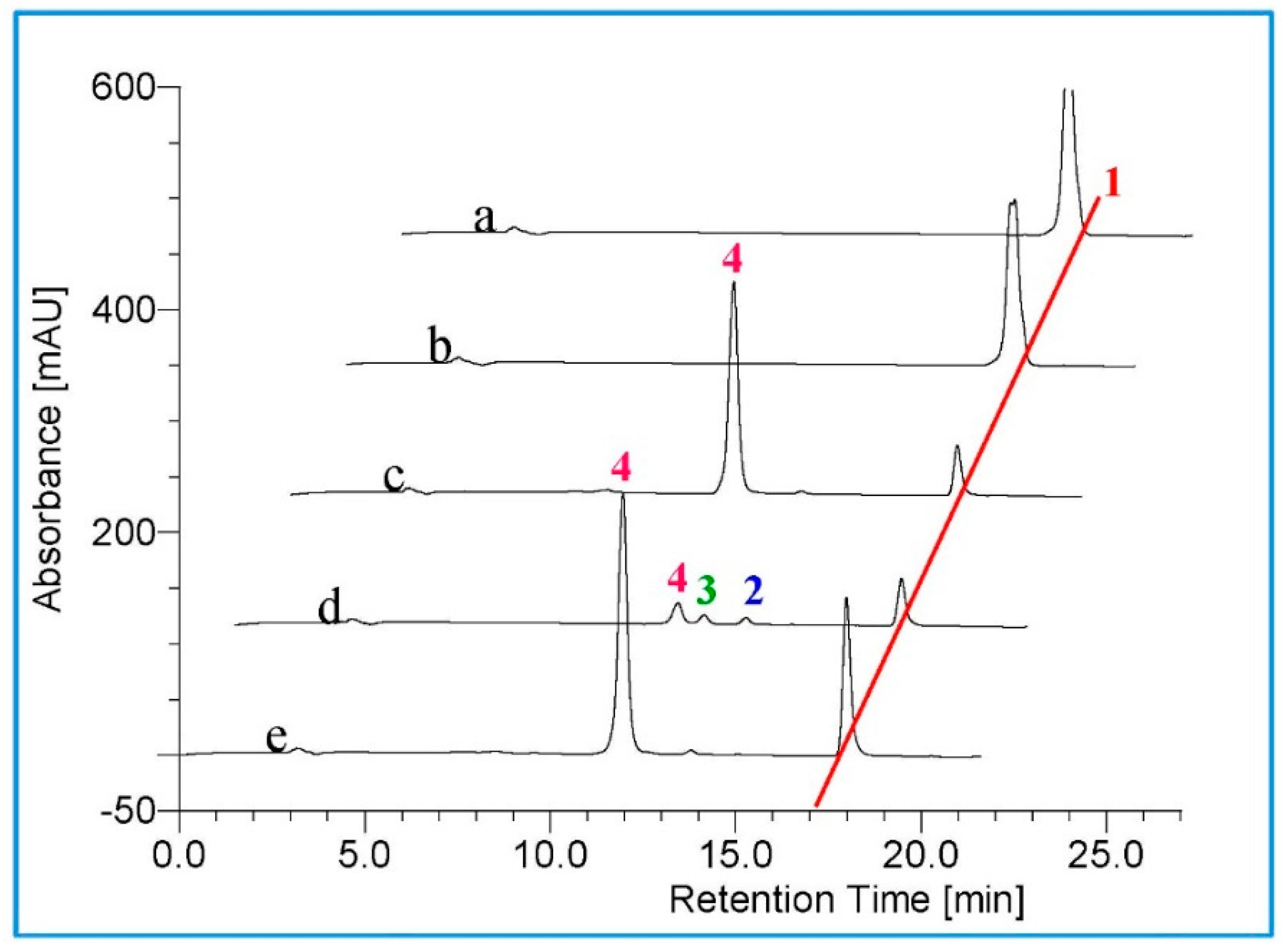
2.2. Metabolism of TES by CYP102A1 Mutants
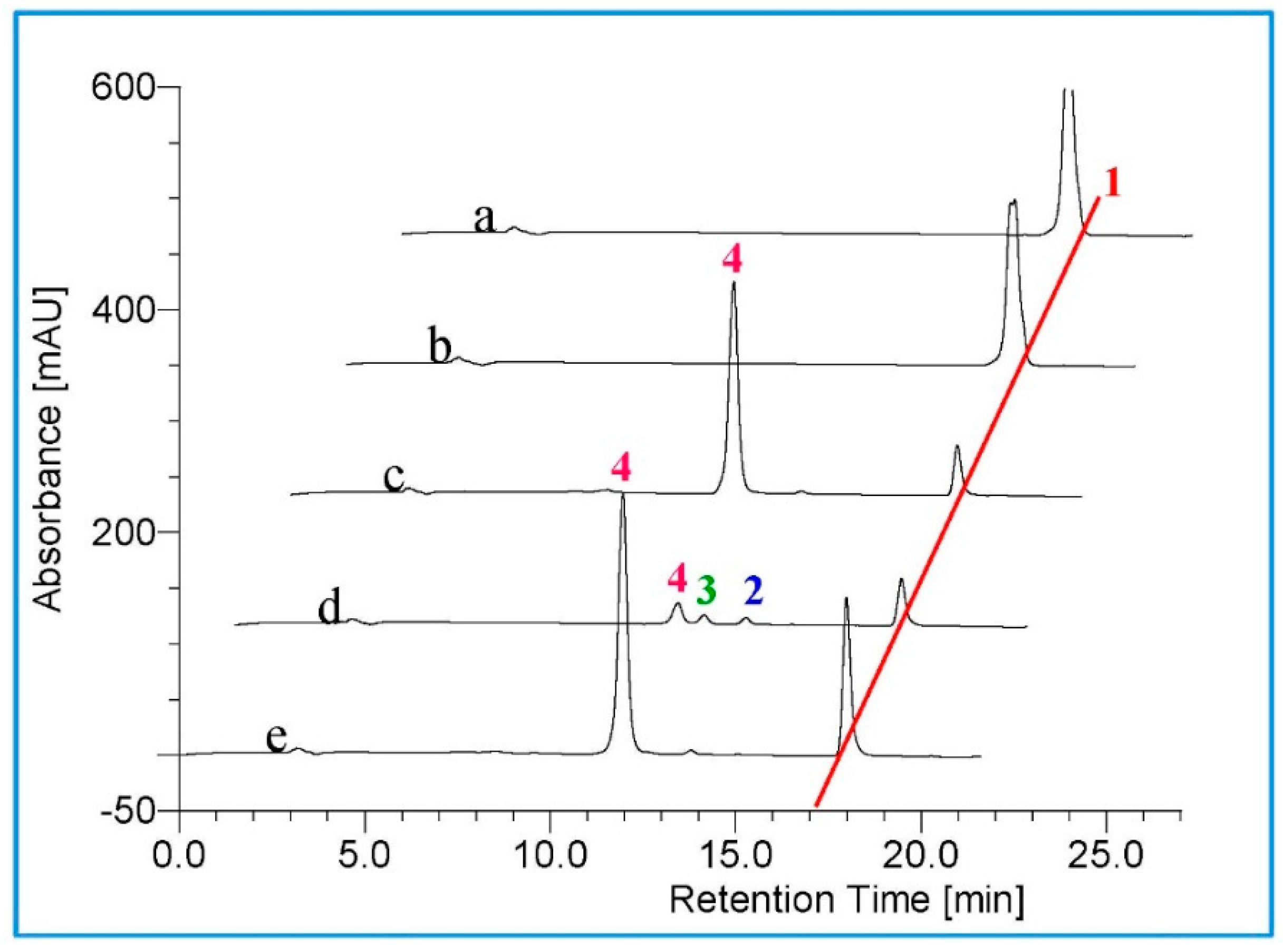
2.3. Metabolism of MT by CYP102A1 Mutants
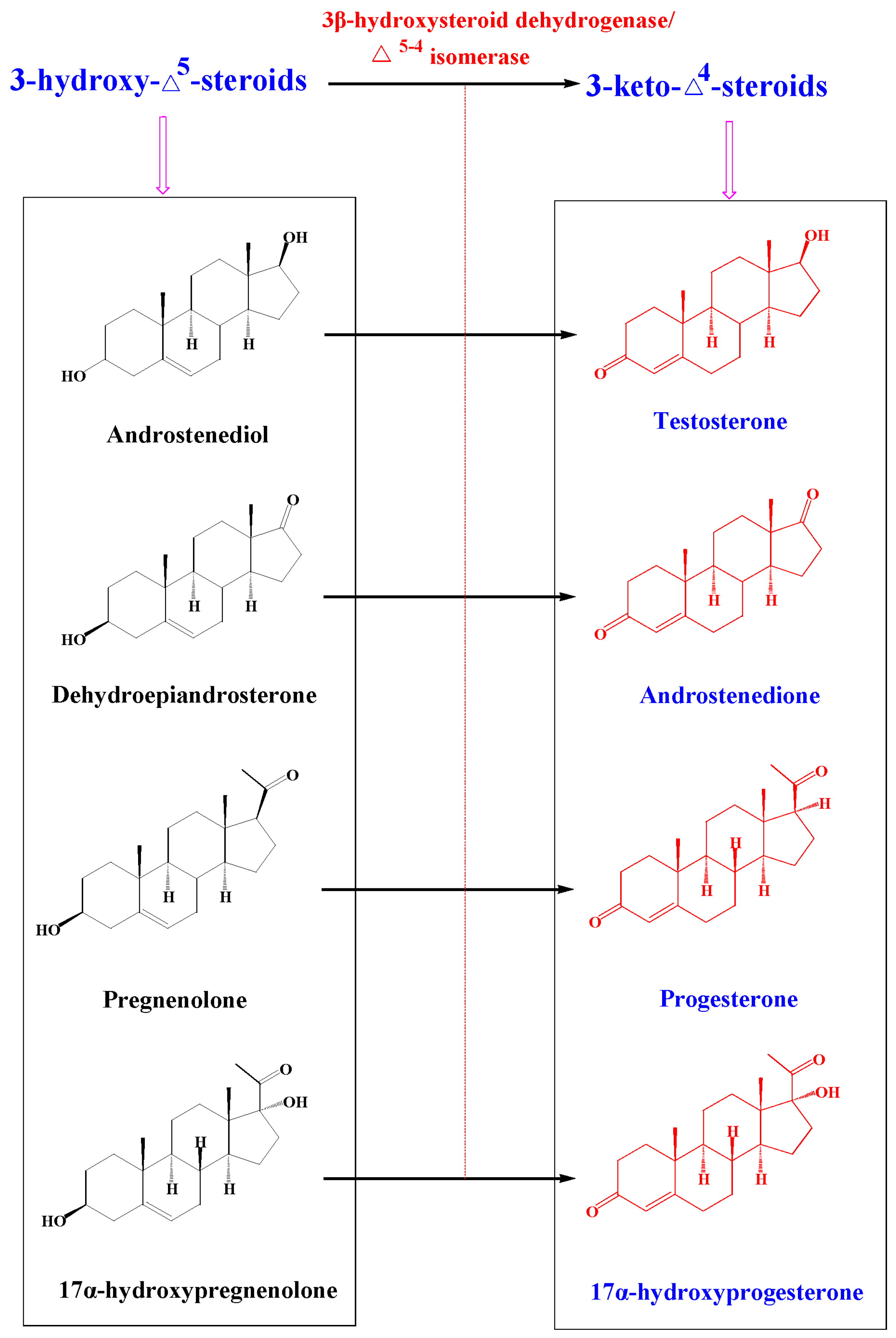
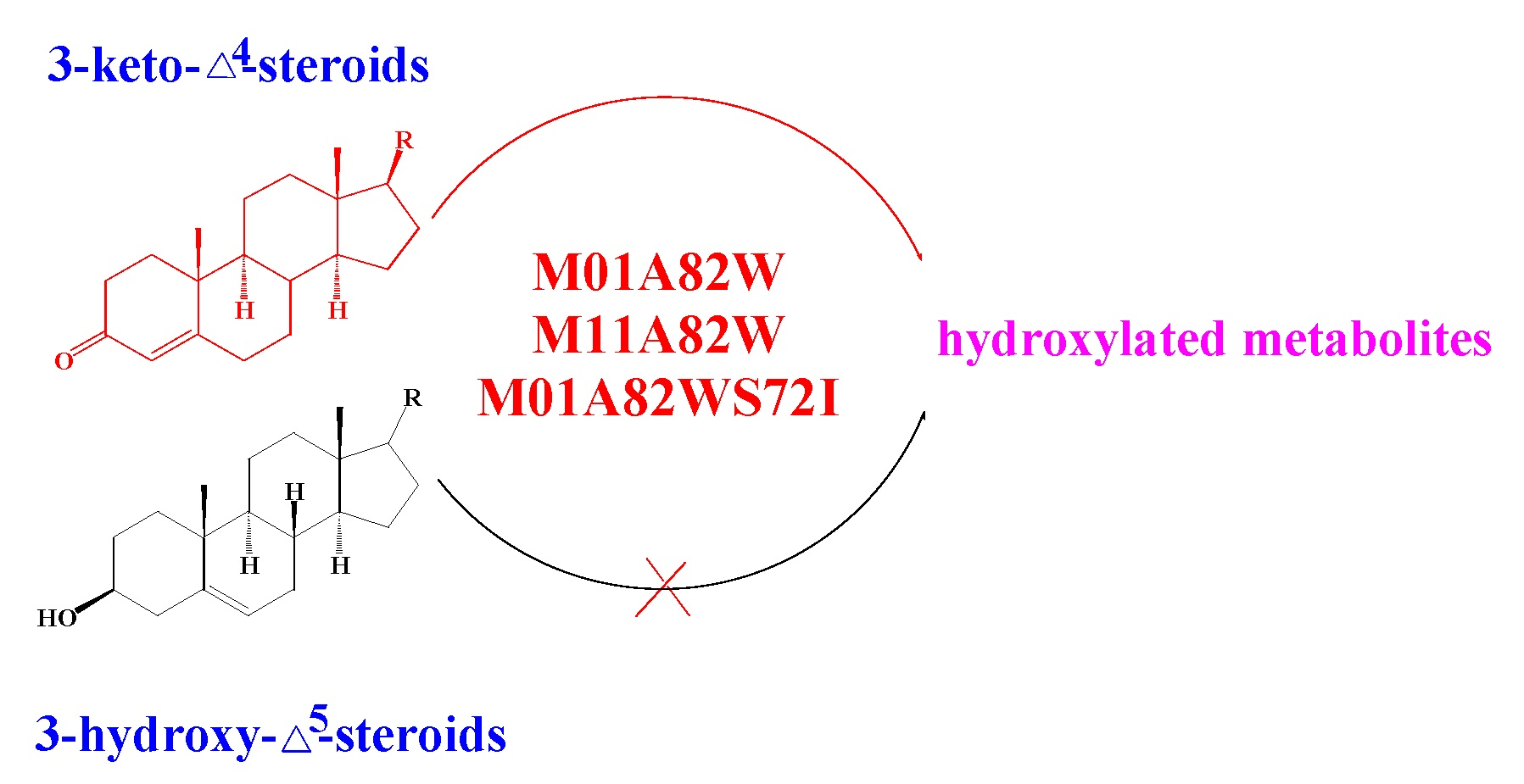
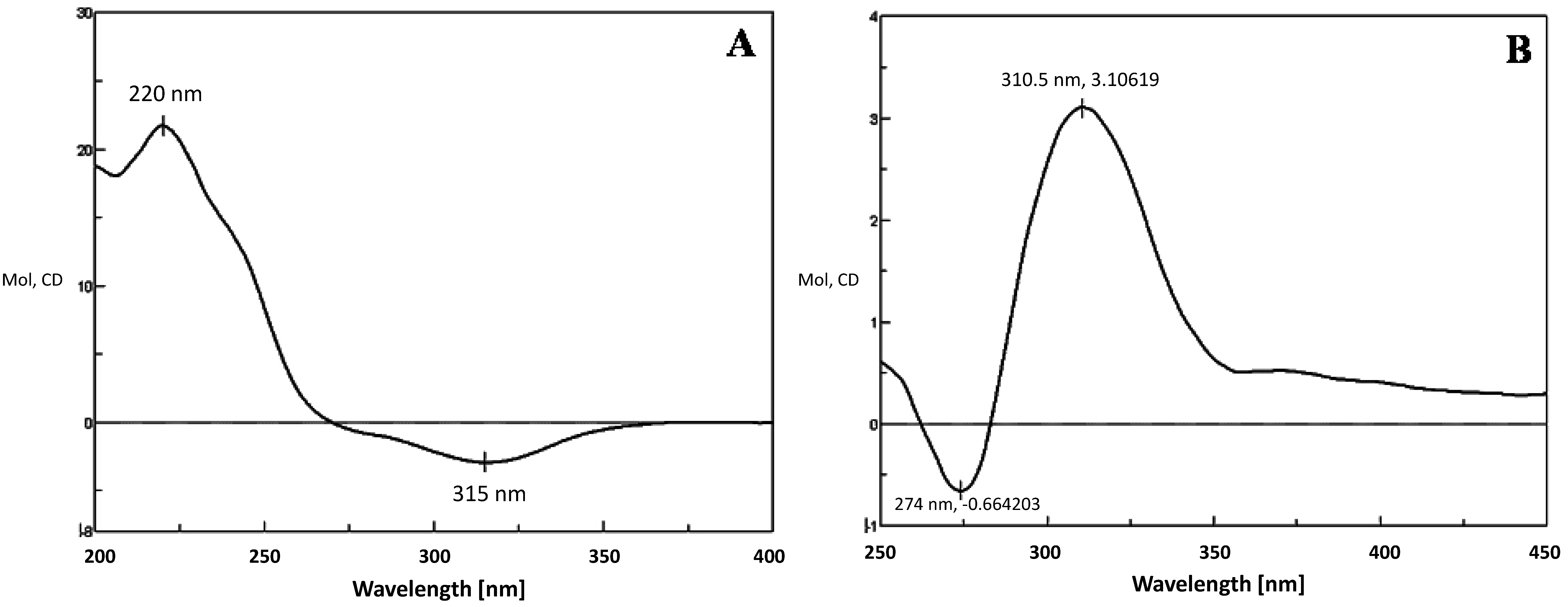
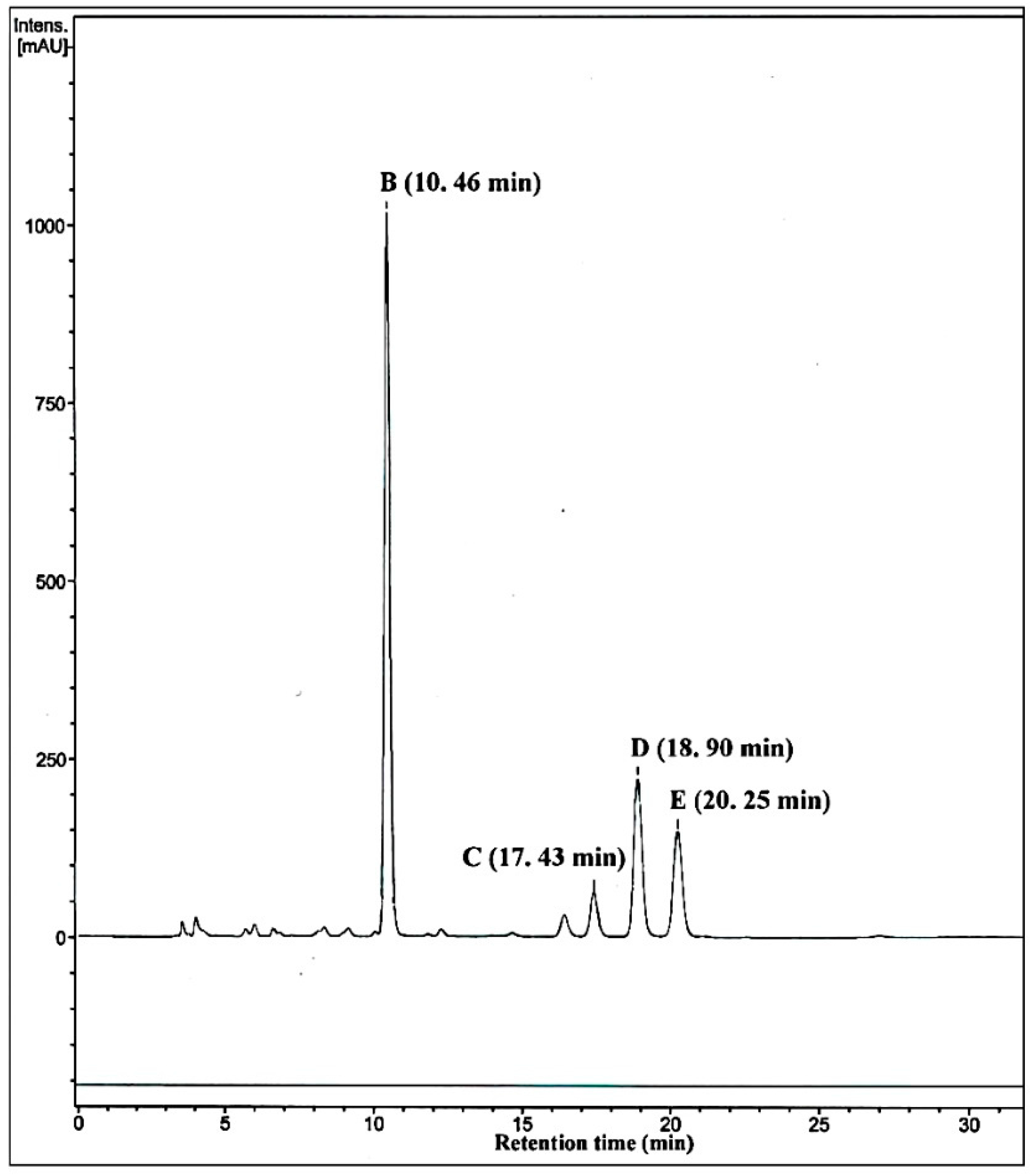
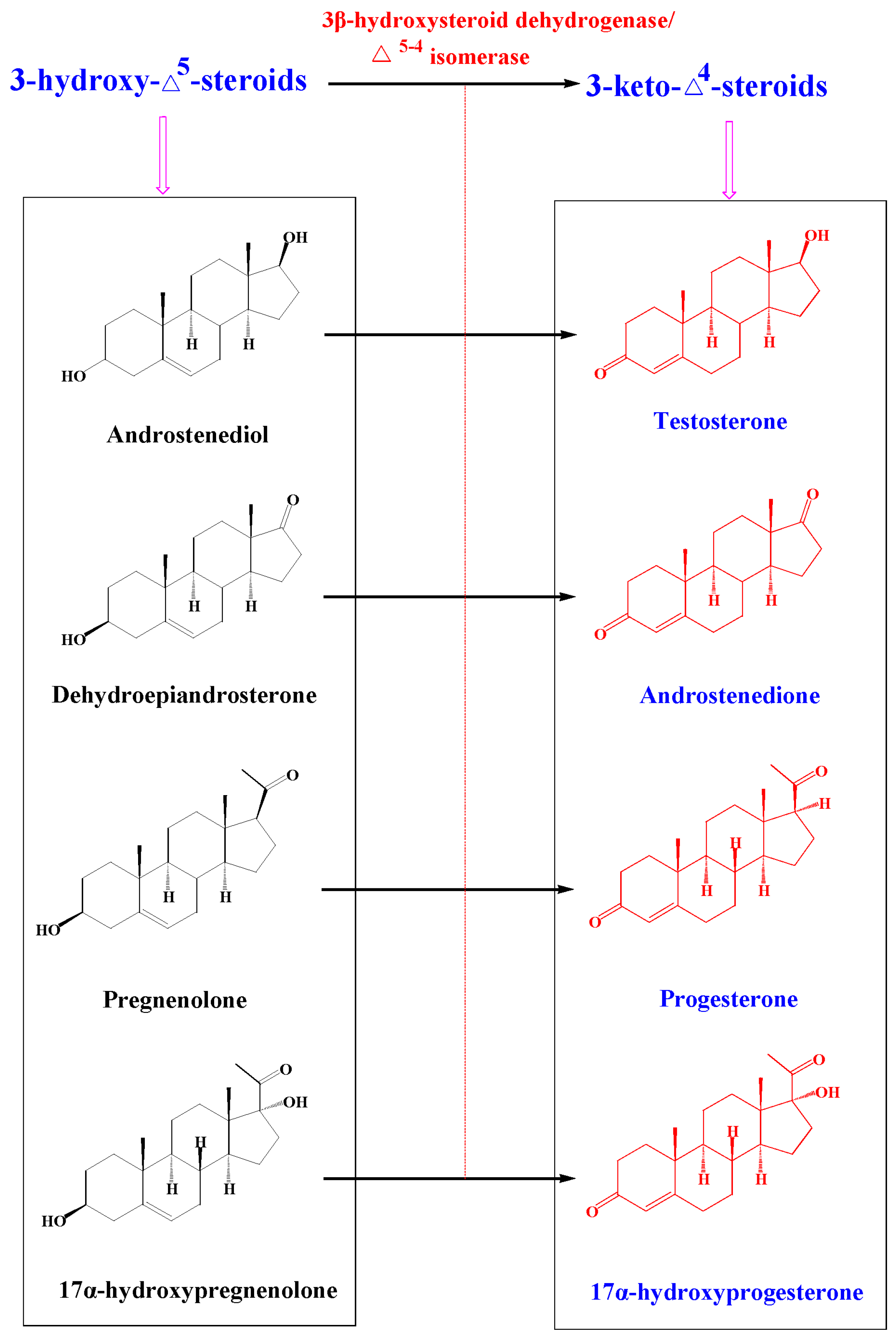
2.4. Metabolism of 3-keto-Δ4-steroids by CYP102A1 Mutants
3. Experimental Section
3.1. Substrates, Chemicals and Enzymes
3.2. Strains and Plasmids
3.3. Site-Directed Mutagenesis
3.4. Expression and Purification of BM3 Mutants
3.5. Metabolism of Steroids by CYP102A1 Mutants
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mimaki, Y.; Kuroda, M.; Kameyama, A.; Sashida, Y.; Hirano, T.; Oka, K.; Maekawa, R.; Wada, T.; Sugita, K.; Beutler, J.A. Cholestane glycosides with potent cytostatic activities on various tumor cells from Ornithogalum saundersiae bulbs. Bioorg. Med. Chem. Lett. 1997, 7, 633–636. [Google Scholar] [CrossRef]
- Kubo, S.; Mimaki, Y.; Terao, M.; Sashida, Y.; Nikaido, T.; Ohmoto, T. Acylated cholestane glycosides from the bulbs of Ornithogalum saundersiae. Phytochemistry 1992, 31, 3969–3973. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kanda, F.; Damarla, S.R.; Rao, D.V.; Rao, C.B. Marine sterols. XVII.: Polyhydroxysterols of the soft corals of the andaman and nicobar coasts.: (2). isolation and structures of three 16β-hydroxy steroidal glycosides from an Alcyonium sp. soft coral. Chem. Pharm. Bull. 1990, 38, 2400–2403. [Google Scholar] [CrossRef]
- Goto, G.; Yoshioka, K.; Hiraga, K.; Masuoka, M.; Nakayama, R.; Miki, T. Synthesis and antiandrogenic activity of 16β-substituted-17β-hydroxysteroids. Chem. Pharm. Bull. 1978, 26, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Löken, B.; Kaufmann, S.; Rosenkranz, G.; Sondheimer, F. Steroids. LXXVII. 1 Synthesis and reactions of 16β-oxygenated pregnan-20-one derivatives. J. Am. Chem. Soc. 1956, 78, 1738–1741. [Google Scholar] [CrossRef]
- Venkataraman, H.; Beer, S.B.; Bergen, L.A.; Essen, N.; Geerke, D.P.; Vermeulen, N.P.; Commandeur, J.N. A single active site mutation inverts stereoselectivity of 16-hydroxylation of testosterone catalyzed by engineered cytochrome P450 BM3. ChemBioChem 2012, 13, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Rea, V.; Kolkman, A.J.; Vottero, E.; Stronks, E.J.; Ampt, K.A.; Honing, M.; Vermeulen, N.P.; Wijmenga, S.S.; Commandeur, J.N. Active site substitution A82W improves the regioselectivity of steroid hydroxylation by cytochrome P450 BM3 mutants as rationalized by spin relaxation nuclear magnetic resonance studies. Biochemistry 2012, 51, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt-Lussenburg, B.M.; Damsten, M.C.; Maasdijk, D.M.; Vermeulen, N.P.; Commandeur, J.N. Heterotropic and homotropic cooperativity by a drug-metabolising mutant of cytochrome P450 BM3. Biochem. Biophys. Res. Commun. 2006, 346, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Pouzar, V.R.; Černý, I.; Lapčı́k, O.; Hill, M.; Hampl, R. Synthesis of two new haptens of 16α-hydroxydehydroepiandrosterone (3β,16α-dihydroxyandrost-5-en-17-one). Steroids 2003, 68, 149–158. [Google Scholar] [CrossRef]
- Denancé, M.; Guyot, M.; Samadi, M. Short synthesis of 16β-hydroxy-5α-cholestane-3, 6-dione a novel cytotoxic marine oxysterol. Steroids 2006, 71, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, H.; Te Poele, E.M.; Rosloniec, K.Z.; Vermeulen, N.; Commandeur, J.N.; van der Geize, R.; Dijkhuizen, L. Biosynthesis of a steroid metabolite by an engineered Rhodococcus erythropolis strain expressing a mutant cytochrome P450 BM3 enzyme. Appl. Microbiol. Biotechnol. 2015, 99, 4713–4721. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.-L. P450BM3 on steroids: the swiss army knife p450 enzyme just gets better. ChemBioChem 2011, 12, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Warman, A.J.; Roitel, O.; Neeli, R.; Girvan, H.M.; Seward, H.E.; Murray, S.A.; McLean, K.J.; Joyce, M.G.; Toogood, H.; Holt, R.A.; et al. Flavocytochrome P450 BM3: An update on structure and mechanism of a biotechnologically important enzyme. Biochem. Soc. Trans. 2005, 33, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.O.; Fulco, A.J. Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 1986, 261, 7160–7169. [Google Scholar] [PubMed]
- Glieder, A.; Farinas, E.T.; Arnold, F.H. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 2002, 20, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, A.B.; Wong, L.-L. Protein engineering of Bacillus megaterium CYP102. Eur. J. Biochem. 2001, 268, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Schwaneberg, U.; Fischer, P.; Schmid, R.D. Directed evolution of the fatty-acid hydroxylase P450 BM-3 into an indole-hydroxylating catalyst. Chem. A Eur. J. 2000, 6, 1531–1536. [Google Scholar] [CrossRef]
- Vottero, E.; Rea, V.; Lastdrager, J.; Honing, M.; Vermeulen, N.P.E.; Commandeur, J.N.M. Role of residue 87 in substrate selectivity and regioselectivity of drug-metabolizing cytochrome P450 CYP102A1 M11. J. Biol. Inorg. Chem. 2011, 16, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Reinen, J.; Vredenburg, G.; Klaering, K.; Vermeulen, N.P.E.; Commandeur, J.N.M.; Honing, M.; Vos, J.C. Selective whole-cell biosynthesis of the designer drug metabolites 15- or 16-betahydroxynorethisterone by engineered Cytochrome P450 BM3 mutants. J. Mol. Catal. B Enzym. 2015, 121, 64–74. [Google Scholar] [CrossRef]
- Kille, S.; Zilly, F.E.; Acevedo, J.P.; Reetz, M.T. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011, 3, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, L.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2 (OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [PubMed]
- Górecki, M.; Jablonska, E.; Kruszewska, A.; Suszczynska, A.; Urbanczyk-Lipkowska, Z.; Gerards, M.; Morzycki, J.W.; Szczepek, W.J.; Frelek, J. Practical method for the absolute configuration assignment of tert/tert 1,2-diols using their complexes with Mo2 (OAc)4. J. Org. Chem. 2007, 72, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Musharraf, S.G.; Ali, A.; Khan, N.T.; Yousuf, M.; Choudhary, M.I. Tandem mass spectrometry approach for the investigation of the steroidal metabolism: Structure–fragmentation relationship (SFR) in anabolic steroids and their metabolites by ESI-MS/MS analysis. Steroids 2013, 78, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Agematu, H.; Matsumoto, N.; Fujii, Y.; Kabumoto, H.; Doi, S.; Machida, K.; Ishikawa, J.; Arisawa, A. Hydroxylation of testosterone by bacterial cytochromes P450 using the Escherichia coli expression system. Biosci. Biotechnol. Biochem. 2006, 70, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-B.; Chen, X.; Wang, W.; Cheng, K.-D.; Kong, J.-Q. Transcriptome-wide identification and characterization of Ornithogalum saundersiae phenylalanine ammonia lyase gene family. RSC Adv. 2014, 4, 27159–27175. [Google Scholar] [CrossRef]
- Li, L.-N.; Kong, J.Q. Transcriptome-wide identification of sucrose synthase genes in Ornithogalum caudatum. RSC Adv. 2016, 6, 18778–18792. [Google Scholar] [CrossRef]
- Guo, L.; Chen, X.; Li, L.-N.; Tang, W.; Pan, Y.-T.; Kong, J.-Q. Transcriptome-enabled discovery and functional characterization of enzymes related to (2S)-pinocembrin biosynthesis from Ornithogalum caudatum and their application for metabolic engineering. Microb. Cell Fact. 2016, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kong, J.-Q. cDNA cloning and expression analysis of farnesyl pyrophosphate synthase from Ornithogalum saundersiae. Z. Naturforsch. C 2014, 69, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Pongtharangkul, T.; Chuekitkumchorn, P.; Suwanampa, N.; Payongsri, P.; Honda, K.; Panbangred, W. Kinetic properties and stability of glucose dehydrogenase from Bacillus amyloliquefaciens SB5 and its potential for cofactor regeneration. AMB Express 2015, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Kong, J.-Q. Transcriptome-guided discovery and functional characterization of two UDP-sugar 4-epimerase families involved in the biosynthesis of anti-tumor polysaccharides in Ornithogalum caudatum. RSC Adv. 2016, 6, 37370–37384. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2β-, 16α- and 16β-OH-T, 2β-, 16α- and 16β-OH-MT and 1α-OH-androstenedione are available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 16β-OH-T | 16α-OH-T * | 2β-OH-T | ||
|---|---|---|---|---|---|
| δC | δH | δH | δC | δH | |
| 1 | 35.7 | 1.50–1.42 (m, 1H); 1.88–1.84 (m, 1H) | 1.50–1.47 (m, 1H); 1.80–1.77 (m, 1H) | 41.3 | 1.63–1.56 (m, 1H); 2.05 (m, 1H) |
| 2 | 34 | 2.06–2.00 (m, 1H); 2.34–2.27 (m, 1H) | 2.04–2.00 (m, 1H); 2.35–2.29 (m, 1H) | 68.6 | 4.00–3.95 (m, 1H) |
| 3 | 199.5 | 199.4 | |||
| 4 | 124 | 5.73 (s, 1H) | 5.72 (s, 1H) | 120.3 | 5.63 (s, 1H) |
| 5 | 170.9 | 172 | |||
| 6 | 32.7 | 2.28–2.26 (m, 1H); 2.37–2.34 (m, 1H) | 2.29–2.25 (m, 1H); 2.42–2.37 (m, 1H) | 30.3 | 2.19–2.16 (m, 1H); 2.40–2.37 (m, 1H) |
| 7 | 31.7 | 1.02 -0.97 (m, 1H); 1.73–1.68 (m, 1H) | 1.00–0.98 (m, 1H); 1.73–1.68 (m, 1H) | 32.5 | 0.85 (m, 1H); 1.87–1.84 (m, 1H) |
| 8 | 35 | 1.50–1.42 (m, 1H) | 1.50–1.47 (m, 1H) | 34.1 | 1.63–1.56 (m, 1H) |
| 9 | 54.1 | 0.97–0.93 (m, 1H) | 0.99–0.96 (m, 1H) | 51.1 | 1.37–1.34 (m, 1H) |
| 10 | 42.4 | 43.3 | |||
| 11 | 20.4 | 1.31–1.27 (m, 1H); 1.35–1.31 (m, 1H) | 1.47–1.44 (m, 2H) | 22.1 | 1.63–1.56 (m, 1H); 1.50–1.44 (m, 1H) |
| 12 | 37 | 1.06–1.03 (m, 1H); 1.67–1.63 (m, 1H) | 1.04–1.00 (m, 1H); 1.43–1.40 (m, 1H) | 35.6 | 1.03 (m, 1H); 1.78–1.72 (m, 1H) |
| 13 | 38.7 | 36.7 | |||
| 14 | 47 | 0.81–0.79 (m, 1H) | 0.81–0.79 (m, 1H) | 50.1 | 0.85 (m, 1H) |
| 15 | 35 | 1.15–1.10 (m, 1H); 1.93–1.88 (m, 1H) | 1.83–1.81 (m, 1H); 1.06–1.04 (m, 1H) | 23.5 | 1.50–1.44 (m, 1H); 1.34 (m, 1H) |
| 16 | 70 | 3.39–3.36 (m, 1H) | 4.17–4.13 (m, 1H) | 29.5 | 1.85–1.81 (m, 1H); 1.43–1.39 (m, 1H) |
| 17 | 80.7 | 4.19 (m, 1H) | 3.50 (d, J 5.7, 1H) | 80.3 | 3.47–3.41 (m, 1H) |
| 18 | 11.9 | 0.86 (s, 3H) | 0.84 (s, 3H) | 11.8 | 0.67 (s, 3H) |
| 19 | 17.4 | 1.20 (s, 3H) | 1.18 (s, 3H) | 22.4 | 1.14 (s, 3H) |
| Position | 16α-OH-MT | 16β-OH-MT | 2β-OH-MT | ||||||
|---|---|---|---|---|---|---|---|---|---|
| δC | δH | HMBC | δC | δH | HMBC | δC | δH | HMBC | |
| 1 | 35.8 | 1.87, m | C2, C3, C10, C19 | 36.8 | 2.16, m | C2, C3 | 41.6 | 1.61–1.68, m | C2, C9, C10, C19 |
| 2 | 33.2 | 2.25, m; 2.42, m | C1, C3, C4, C10 | 33.9 | 2.25, m; 2.43, m | C1, C3, C4 | 69.8 | 4.15–4.18, m | C1, C3 |
| 3 | 200.9 | 202.4 | 201.6 | ||||||
| 4 | 122.7 | 5.68, s | C3, C5 | 124.1 | 5.68,s | C3, C5 | 120.5 | 5.69, s | C2, C6, C10 |
| 5 | 173.7 | 175.1 | 174.3 | ||||||
| 6 | 33.2 | 2.28, m; 2.46, m | C4, C5, C7 | 34.7 | 2.29, m; 2.47, m | C4, C5, C7 | 32.7 | 2.39–2.45, m;2.51–2.59, m | C5, C7, C10 |
| 7 | 31.4 | 1.38, m; 1.81, m | C6, C8, C9 | 33.1 | 1.03, m;1.87, m | C6, C8, C9 | 33.9 | 1,31, m | C5, C9 |
| 8 | 35.8 | 1.60, m | C6, C8, C9 | 37.2 | 1.69, m | C7, C9 | 35.7 | 1.61–1.68, m | C9, C10, C14, C15 |
| 9 | 35.8 | 0.93, m | C8, C10, C11, C12, C19 | 55.4 | 0.91, m | C8, C10, C11, C19 | 51.5 | 0.93, m | C8, C10 |
| 10 | 38.6 | 37.2 | 42.4 | ||||||
| 11 | 19.8 | 1.46, m; 1.51, m | C9, C12 | 21.6 | 1.48, m;1.53, m | C9, C12 | 27.1 | 1.51–1.56, m; 1.61–1.68, m | C9, C12 |
| 12 | 31.5 | 1.00, m; 1.67, m | C9, C18 | 33.5 | 0.94, m; 1.60, m | C9, C13, C18 | 37.8 | 1.26–1.28, m | C18 |
| 13 | 45.8 | 46.0 | 47.2 | ||||||
| 14 | 47.9 | 1.41, m | C13, C15, C16, C18 | 47.9 | 1.31, m | C13, C15, C16, C18 | 49.9 | 0.95–1.03, m | C8, C9, C15 |
| 15 | 32.4 | 1.35, m; 2.06, m | C13, C14, C16 | 35.8 | 0.99, m;2.06m | C13, C14, C16 | 24.2 | 1.28–1.30, m; 1.51–1.56, m | C14, C16 |
| 16 | 79.1 | 4.15, m | C14, C15, C17 | 78.3 | 3.58, m | C15, C17, C20 | 26.1 | 1.80–188, m | C15 |
| 17 | 83.4 | 79.8 | 82.0 | ||||||
| 18 | 13.4 | 0.89, s | C12, C13, C17 | 14.2 | 0.87, s | C12, C13, C17 | 14.7 | 0.87, s | C12, C13, C17 |
| 19 | 16.2 | 1.21, s | C1, C9 | 17.7 | 1.22, s | C9, C10 | 23.0 | 1.21, s | C13, C16, C17 |
| 20 | 16.7 | 1.11, s | C13, C16, C17 | 24.2 | 1.07, s | C13, C16, C17 | 23.5 | 1.16, s | C5, C9, C10 |
| Position | 1α-OH-Androstenedione | |
|---|---|---|
| δC | δH | |
| 1 | 74.77 | 3.81 (t, J = 8.6 Hz, 1H) |
| 2 | 35.73 | 2.40–2.36 (m, 1H) 2.42 (dd, J = 14.0 Hz, 5.0, 1H) |
| 3 | 198.22 | |
| 4 | 123.55 | 5.57 (s, 1H) |
| 5 | 170.74 | |
| 6 | 32.34 | 2.26–2.19 (m, 2H) |
| 7 | 31.59 | 0.94-0.88 (m, 1H) 1.88–1.85 (m, 1H) |
| 8 | 33.96 | 1.58–1.53 (m, 1H) |
| 9 | 46.63 | 1.68–1.63 (m, 1H) |
| 10 | 38.92 | |
| 11 | 20.3 | 1.43–1.34 (m, 1H )1.58–1.53 (m, 1H) |
| 12 | 31.85 | 1.02–0.95 (m, 1H) 1.74 (ddd, J = 22.3, 11.2, 3.5 Hz, 1H) |
| 13 | 44.81 | |
| 14 | 54.24 | |
| 15 | 31.24 | 1.43–1.34 (m, 1H) 1.96–1.92 (m, 1H) |
| 16 | 35.52 | 2.13–2.07 (m, 1H) 2.36–2.30 (m, 1H) |
| 17 | 218.95 | |
| 18 | 14.16 | 0.85 (s, 3H) |
| 19 | 16.7 | 1.15 (s, 3H) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Wang, Z.-B.; Wang, Y.-N.; Kong, J.-Q. Probing Steroidal Substrate Specificity of Cytochrome P450 BM3 Variants. Molecules 2016, 21, 760. https://doi.org/10.3390/molecules21060760
Liu X, Wang Z-B, Wang Y-N, Kong J-Q. Probing Steroidal Substrate Specificity of Cytochrome P450 BM3 Variants. Molecules. 2016; 21(6):760. https://doi.org/10.3390/molecules21060760
Chicago/Turabian StyleLiu, Xing, Zhi-Biao Wang, Ya-Nan Wang, and Jian-Qiang Kong. 2016. "Probing Steroidal Substrate Specificity of Cytochrome P450 BM3 Variants" Molecules 21, no. 6: 760. https://doi.org/10.3390/molecules21060760
APA StyleLiu, X., Wang, Z. -B., Wang, Y. -N., & Kong, J. -Q. (2016). Probing Steroidal Substrate Specificity of Cytochrome P450 BM3 Variants. Molecules, 21(6), 760. https://doi.org/10.3390/molecules21060760