The In Vitro Effects of Pentamidine Isethionate on Coagulation and Fibrinolysis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents, Enzymes, and Substrates
2.2. In vitro Effect on Clotting Times of Human Plasmas
2.3. In Vitro Effect on Fibrinolysis
2.4. Effect of Pentamidine Isethionate on Common Coagulation Pathway and Fibrinolysis Enzymes
2.5. Effect of Pentamidine Isethionate on Related Enzymes
2.6. Michaelis-Menten Kinetics for Chromogenic Substrates Hydrolysis by FXa and Plasmin in the Presence of Pentamidine Isethionate
2.7. Molecular Modeling of Pentamidine Binding to the Active Site of Human FXa and Plasmin
3. Results
3.1. Pentamidine Affects the Clotting Times of Human Plasma and the Effect Is Independent of Factors VIIa, IXa, XIa, and XIIa
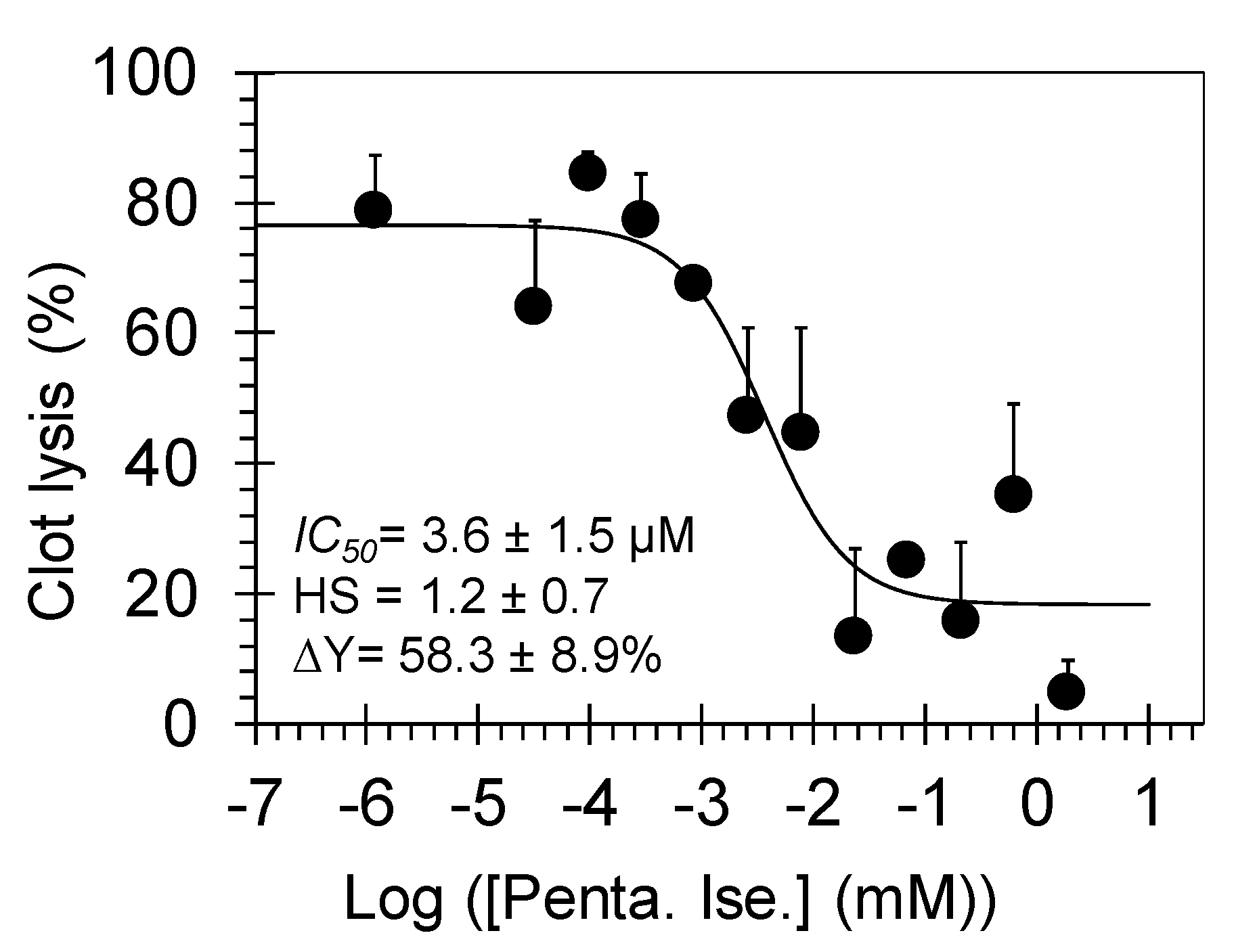
3.2. Pentamidine Dose-Dependently Inhibits Plasmin-Mediated Clot Lysis
3.3. Pentamidine Isethionate Selectively Inhibits Human FXa and Plasmin in the Chromogenic Substrate Hydrolysis Assays
3.4. Michaelis-Menten Kinetics of FXa and Plasmin Inhibition by Pentamidine Isethionate
3.5. Molecular Modeling Studies of Pentamidine Interaction with FXa and Plasmin
4. Discussion and Significance
Author Contributions
Funding
Conflicts of Interest
References
- Khaw, M.; Panosian, C.B. Human antiprotozoal therapy: Past, present, and future. Clin. Microbiol. Rev. 1995, 8, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Goa, K.L.; Campoli-Richards, D.M. Pentamidine isethionate. A review of its antiprotozoal activity, pharmacokinetic properties and therapeutic use in Pneumocystis carinii pneumonia. Drugs 1987, 33, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Pohlig, G.; Bernhard, S.C.; Blum, J.; Burri, C.; Mpanya, A.; Lubaki, J.P.; Mpoto, A.M.; Munungu, B.F.; N’tombe, P.M.; Deo, G.K.; et al. Efficacy and safety of pafuramidine versus pentamidine maleate for treatment of first stage sleeping sickness in a randomized, comparator-controlled, international phase 3 clinical trial. PLoS Negl. Trop. Dis. 2016, 10, e0004363. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.G.; Morrison-Bryant, M. Management of Pneumocystis Jirovecii pneumonia in HIV infected patients: Current options, challenges and future directions. HIV AIDS (Auckl.) 2010, 2, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Pentamidine Isethionate. FDA Drug Label. Available online: https://www.accessdata.fda.gov/spl/data/589fcce2-fa9a-4dc0-a79b-ae87cd024365/589fcce2-fa9a-4dc0-a79b-ae87cd024365.xml#s10 (accessed on 12 May 2019).
- Suzuki, T.; Takahashi, Y.; Yoshida, S.; Yamagishi, T.; Tateyama, M.; Tanaka, A.; Koshihara, K.; Matsumoto, K.; Fukue, H.; Yorifuji, H.; et al. HIV-1 seropositive hemophilia A complicated by disseminated intravascular coagulation syndrome and acute pancreatitis during treatment of Pneumocystis carinii pneumonia. Rinsho Ketsueki 1992, 33, 1273–1278. [Google Scholar]
- Kempin, S.J.; Jackson, C.W.; Edwards, C.C. In vitro inhibition of platelet function and coagulation by pentamidine isethionate. Antimicrob. Agents Chemother. 1977, 12, 451–454. [Google Scholar] [CrossRef]
- Obaidullah, A.J.; Al-Horani, R.A. Discovery of chromen-7-yl furan-2-carboxylate as a potent and selective factor XIa inhibitor. Cardiovasc. Hematol. Agents Med. Chem. 2017, 15, 40–48. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Gailani, D.; Desai, U.R. Allosteric inhibition of factor XIa. Sulfated non-saccharide glycosaminoglycan mimetics as promising anticoagulants. Thromb. Res. 2015, 136, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Afosah, D.K.; Al-Horani, R.A.; Sankaranarayanan, N.V.; Desai, U.R. Potent, selective, allosteric inhibition of human plasmin by sulfated non-saccharide glycosaminoglycan mimetics. J. Med. Chem. 2017, 60, 641–657. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Karuturi, R.; White, D.T.; Desai, U.R. Plasmin regulation through allosteric, sulfated, small molecules. Molecules 2015, 20, 608–624. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Ponnusamy, P.; Mehta, A.Y.; Gailani, D.; Desai, U.R. Sulfated pentagalloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J. Med. Chem. 2013, 56, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Kummarapurugu, A.B.; Afosah, D.K.; Sankaranarayanan, N.V.; Navaz Gangji, R.; Zheng, S.; Kennedy, T.; Rubin, B.K.; Voynow, J.A.; Desai, U.R. Molecular principles for heparin oligosaccharide-based inhibition of neutrophil elastase in cystic fibrosis. J. Biol. Chem. 2018, 293, 12480–12490. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Karuturi, R.; Lee, M.; Afosah, D.K.; Desai, U.R. Allosteric inhibition of factor XIIIa. Non-saccharide glycosaminoglycan mimetics, but not glycosaminoglycans, exhibit promising inhibition profile. PLoS ONE 2016, 11, e0160189. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Tsai, Y.H. A rapid transglutaminase assay for high-throughput screening applications. J. Biomol. Screen. 2006, 11, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2015-3: Glide; Schrödinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Brandstetter, H.; Kühne, A.; Bode, W.; Huber, R.; von der Saal, W.; Wirthensohn, K.; Engh, R.A. X-ray structure of active site-inhibited clotting factor Xa. Implications for drug design and substrate recognition. J. Biol. Chem. 1996, 271, 29988–29992. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.A.; Fernandez-Catalan, C.; Bergner, A.; Huber, R.; Hopfner, K.P.; Schlott, B.; Gührs, K.H.; Bode, W. The ternary microplasmin-staphylokinase-microplasmin complex is a proteinase-cofactor-substrate complex in action. Nat. Struct. Biol. 1998, 5, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Afosah, D.K. Recent advances in the discovery and development of factor XI/XIa inhibitors. Med. Res. Rev. 2018, 38, 1974–2023. [Google Scholar] [CrossRef]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer, J.; Schlemmer, K.H.; Reinemer, P.; Perzborn, E. Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide (BAY 59-7939): An oral, direct factor Xa inhibitor. J. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef]
- Samama, M.M.; Martinoli, J.L.; LeFlem, L.; Guinet, C.; Plu-Bureau, G.; Depasse, F.; Perzborn, E. Assessment of laboratory assays to measure rivaroxaban—An oral, direct factor Xa inhibitor. Thromb. Haemost. 2010, 103, 815–825. [Google Scholar] [PubMed]
- Baglin, T.; Keeling, D.; Kitchen, S. British Committee for Standards in Haematology. Effects on routine coagulation screens and assessment of anticoagulant intensity in patients taking oral dabigatran or rivaroxaban: Guidance from the British Committee for Standards in Haematology. Br. J. Haematol. 2012, 159, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Conte, J.E., Jr.; Upton, R.A.; Phelps, R.T.; Wofsy, C.B.; Zurlinden, E.; Lin, E.T. Use of a specific and sensitive assay to determine pentamidine pharmacokinetics in patients with AIDS. J. Infect. Dis. 1986, 154, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Conte, J.E., Jr.; Upton, R.A.; Lin, E.T. Pentamidine pharmacokinetics in patients with AIDS with impaired renal function. J. Infect. Dis. 1987, 156, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Conte, J.E., Jr. Pharmacokinetics of intravenous pentamidine in patients with normal renal function or receiving hemodialysis. J. Infect. Dis. 1991, 163, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bronner, U.; Gustafsson, L.L.; Doua, F.; Ericsson, O.; Miézan, T.; Rais, M.; Rombo, L. Pharmacokinetics and adverse reactions after a single dose of pentamidine in patients with Trypanosoma gambiense sleeping sickness. Br. J. Clin. Pharmacol. 1995, 39, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.Q.; Campana, P.T.; Melo, P.A.; Bianconi, M.L. Suramin interaction with human alpha-thrombin: inhibitory effects and binding studies. Int. J. Biochem. Cell Biol. 2004, 36, 2077–2085. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Human Plasma | [Pentamidine Isethionate] (µm) to Double Clotting Time |
|---|---|
| APTT assay | APTTEC2× |
| Normal | 27.88 b) ± 2.99 c) |
| Deficient of FVII | 26.71 ± 0.50 |
| Deficient of FIX | 26.32 ± 3.52 |
| Deficient of FXI | 29.48 ± 3.88 |
| Deficient of FXII | 27.05 ± 6.05 |
| PT assay | PTEC2× |
| Normal | 45.70 ± 0.85 |
| TT assay | TTEC2× |
| Normal | 158.17 ± 30.87 |
| Enzyme | IC50 (µM) | HS | ∆Y (%) |
|---|---|---|---|
| Thrombin | >100 b) | NA c) | NA |
| FXa | 10.4 ± 0.8 d) | 0.97 ± 0.07 | 101.6 ± 2.9 |
| FXIa | >100 | NA | NA |
| FXIIIa | >50 | NA | NA |
| Plasmin | 8.4 ± 0.8 | 1.1 ± 0.1 | 98.0 ± 3.3 |
| Neutrophil Elastase | >100 | NA | NA |
| Trypsin | 4.9 ± 0.8 | 1.0 ± 0.2 | 116.8 ± 7.4 |
| Chymotrypsin | >1750 | NA | NA |
| Inhibitor | Spectrozyme FXa | |
| Pentamidine Isethionate (µM) | KM (µM) | VMAX (mAU/min) |
| 0 | 305.0 ± 11.8 b) | 169.4 ± 3.6 |
| 5 | 266.8 ± 21.2 | 114.3 ± 4.7 |
| 10 | 410.3 ± 18.2 | 124.2 ± 3.3 |
| 20 | 364.4 ± 36.6 | 63.1 ± 3.6 |
| 50 | 690.2± 63.1 | 60.3 ± 3.8 |
| Inhibitor | Spectrozyme PL | |
| Pentamidine Isethionate (µM) | KM (µM) | VMAX (mAU/min) |
| 0 | 46.5 ± 5.1 | 76.2 ± 2.5 |
| 10 | 92.9 ± 6.2 | 78.7 ± 2.0 |
| 20 | 176.1 ± 4.3 | 88.4 ± 1.0 |
| 50 | 358.9 ± 21.3 | 92.9 ± 3.2 |
| 100 | 645.5 ± 68.17 | 90.9 ± 6.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Horani, R.A.; Clemons, D.; Mottamal, M. The In Vitro Effects of Pentamidine Isethionate on Coagulation and Fibrinolysis. Molecules 2019, 24, 2146. https://doi.org/10.3390/molecules24112146
Al-Horani RA, Clemons D, Mottamal M. The In Vitro Effects of Pentamidine Isethionate on Coagulation and Fibrinolysis. Molecules. 2019; 24(11):2146. https://doi.org/10.3390/molecules24112146
Chicago/Turabian StyleAl-Horani, Rami A., Daytriona Clemons, and Madhusoodanan Mottamal. 2019. "The In Vitro Effects of Pentamidine Isethionate on Coagulation and Fibrinolysis" Molecules 24, no. 11: 2146. https://doi.org/10.3390/molecules24112146