
Dually Acting Nonclassical 1,4-Dihydropyridines Promote the Anti-Tuberculosis (Tb) Activities of Clofazimine


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
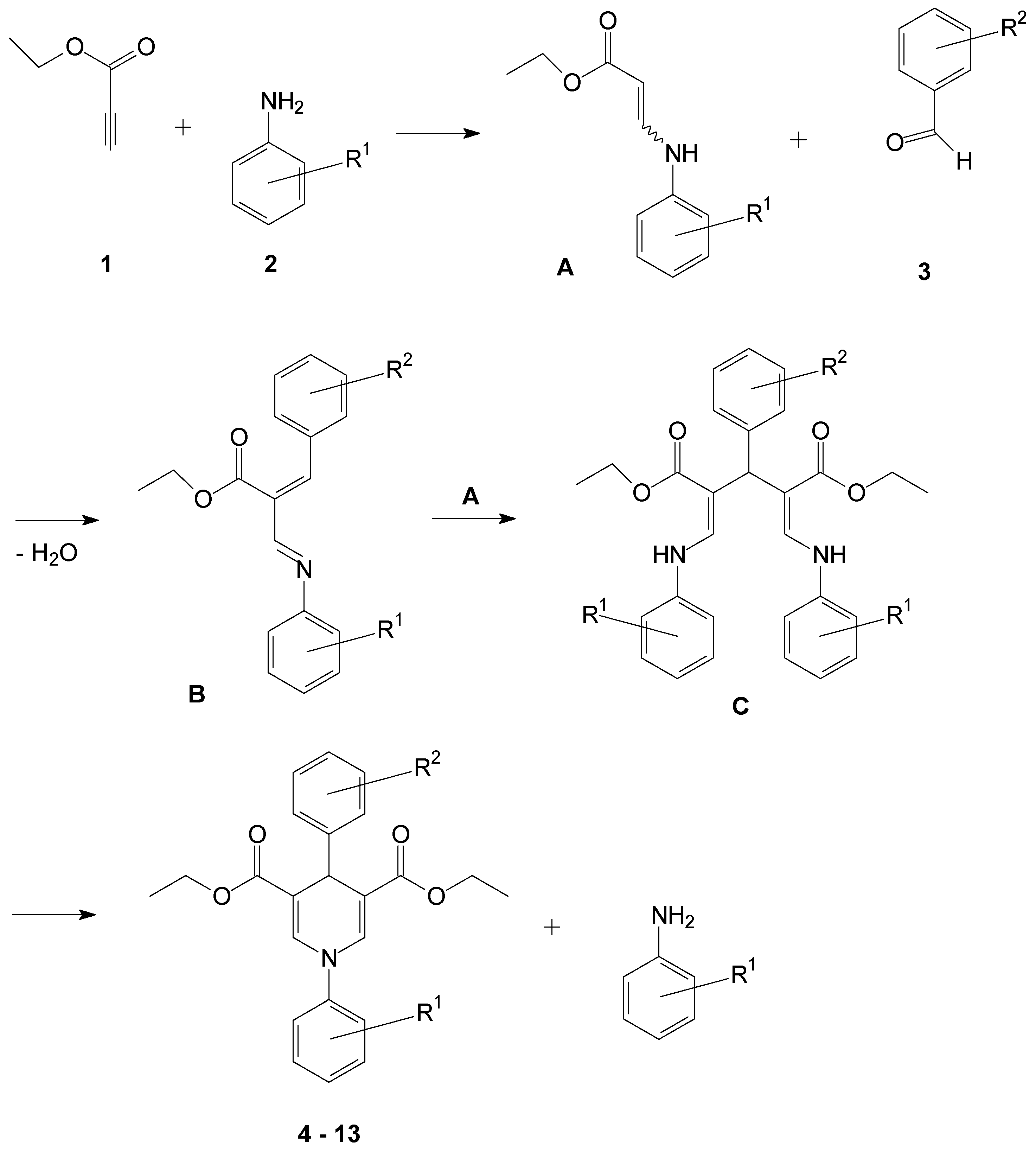
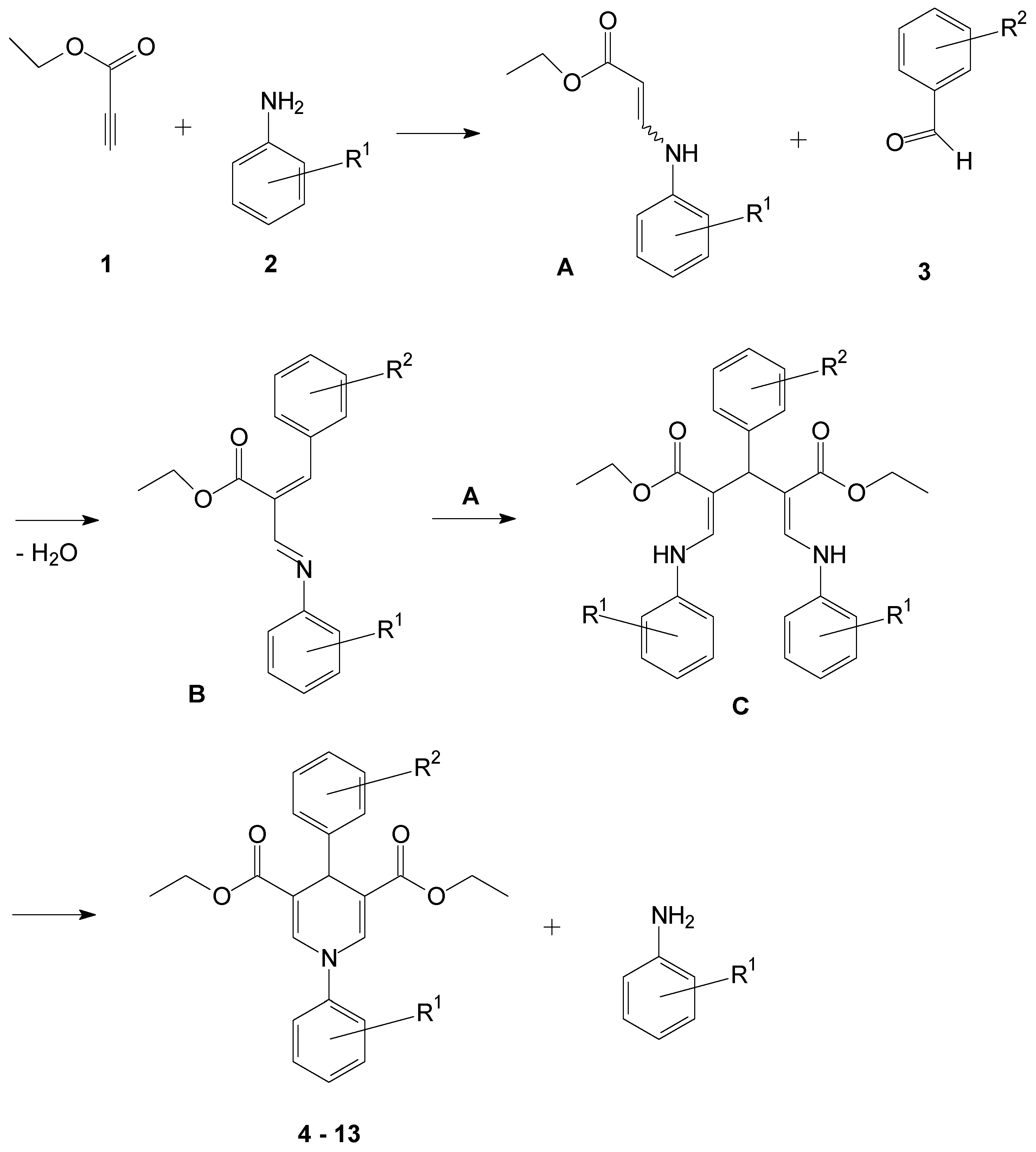
2.1. Synthesis of the 1,4-Dihydropyridines
2.2. Antituberculostatic Activity of the 1,4-Dihydropyridines
2.3. ABCB1 Inhibitory Activity of the 1,4-Dihydropyridines
2.4. Antituberculostatic Drug Toxicity Enhancing Properties of 1,4-Dihydropyridines
3. Material and Methods
3.1. Chemical Reagents and Instruments
3.2. General Procedure for the Synthesis of Compounds 4–18
3.3. ABCB1 Inhibitory Activity
3.4. Mtb Growth Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tiberi, S.; Munoz-Torrico, M.; Duarte, R.; Dalcolmo, M.; D´Ambrosio, L.; Migliori, G.-B. New drugs and perspectives for new anti-tuberculosis regimes. Pulmonology 2018, 24, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2016. Available online: https://apps.who.int/iris/handle/10665/250441 (accessed on 5 August 2019).
- Combs, D.L.; O´Brien, R.J.; Geiter, L.J. USPHS Tuberculosis Short-Course Chemotherapy Trial: Effectiveness, toxicity, and acceptability. The report of final results. Ann. Intern. Med. 1990, 112, 397–406. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guideline for Treatment of Drug-Susceptible Tuberculosis and Patient Care; 2017 update; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Koch, A.; Cox, H.; Mizrahi, V. Drug-resistant tuberculosis: Challenges and opportunities for diagnosis and treatment. Curr. Opin. Pharmacol. 2018, 42, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2017. Available online: https://reliefweb.int/report/world/global-tuberculosis-report-2017 (accessed on 5 August 2019).
- Desai, B.; Sureja, D.; Naliapara, Y.; Shah, A.; Saxena, A.K. Synthesis and QSAR Studies of 4-Substituted Phenyl-2,6-dimethyl-3,5-Bis-N-(substituted Phenyl)carbamoyl-1.4-dihydropyri- dines as Potential Antitubercular Agents. Bioorg. Med. Chem. 2001, 9, 1993–1998. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Skrai, Y. Increased accumulation of vincristine and adriamycin in drug-resistant P388 tumor cells following incubation with calcium antagonists and calmodulin inhibitors. Cancer Res. 1982, 42, 4730–4733. [Google Scholar] [PubMed]
- Hollt, V.; Kouba, M.; Dietel, M.; Vogt, G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem. Pharmacol. 1992, 43, 2601–2608. [Google Scholar] [CrossRef]
- Vijesh, A.M.; Isloor, A.M.; Peethambar, S.K.; Shivananda, K.N.; Arulmoli, T.; Isloor, N.A. Hantzsch reaction: Synthesis and characterization of some new 1,4-dihydropyridine derivatives as potent antimicrobial and antioxidant agents. Eur. J. Med. Chem. 2011, 46, 5591–5597. [Google Scholar] [CrossRef]
- Chennat, T.; Eisner, U. A new synthesis of 1,4-dihydropyridines. J. Chem. Soc. Perkin Trans. 1 1975, 10, 926. [Google Scholar] [CrossRef]
- Yang, J.; Wang, C.; Xie, X.; Li, H.; Li, Y. Acid-Catalyzed Cascade Reactions of Enaminones with Aldehydes. C-H Functionalizations to Afford 1,4-Dihydropyridines. Eur. J. Org. Chem. 2010, 22, 4189–4193. [Google Scholar] [CrossRef]
- Sueki, S.; Takei, R.; Abe, J.; Shimizu, I. Ytterbium-catalyzed synthesis of dihydropyridines. Tetrahedron Lett. 2011, 52, 4473–4477. [Google Scholar] [CrossRef]
- Desai, P.V.; Raub, T.J.; Blanco, M.-J. How hydrogen bonds impact P-glycoprotein transport and permeability. Bioorg. Med. Chem. Lett. 2012, 22, 6540–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelig, A. A general pattern for substrate recognition by P-glycoprotein. Eur. J. Biochem. 1998, 251, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Piddock, L.J. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 2006, 19, 382–402. [Google Scholar] [CrossRef] [PubMed]
- Garima, K.; Pathak, R.; Tandon, R.; Rathor, N.; Sinha, R.; Bose, M.; Varma-Basil, M. Differential expression of efflux pump genes of Mycobacterium tuberculosis in response to varied subinhibitory concentrations of antituberculotic agents. Tuberculosis 2015, 95, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Parish, T.; Balganesh, M.; Ainsa, J.A. Antituberculosis drugs: Reducing efflux = increasing activity. Drug Discov. Today 2017, 22, 592–599. [Google Scholar] [CrossRef] [PubMed]
- O´Donnell, M.R.; Padayatchi, N.; Metcalfe, J.Z. Elucidating the role of clofazimine for the treatment of tuberculosis. Int. J. Tubercol. Lung Dis. 2016, 20, 552–557. [Google Scholar]
- Holdiness, M.R. Clinical pharmacokinetics of clofazimine. A review. Clin. Pharmacokinet. 1989, 16, 74–85. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Treatment Guidelines for Drug-Resistant Tuberculosis; October 2016 Revision; WHO: Geneva, Switzerland, 2016; WHO/HTM/TB/2016.04. [Google Scholar]
- Horita, Y.; Doi, N. Comparative Study of the Effects of Antituberculosis Drugs and Antitretroviral Drugs on Cytochrome P450 3A4 and P-Glycoprotein. Antimicrob. Agents Chemother. 2014, 58, 3168–3176. [Google Scholar] [CrossRef]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldman, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immuneresponsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef]
- Wiebach, V.; Mainz, A.; Siegert, M.J.; Jungmann, N.A.; Lesquame, G.; Tirat, S.; Dreux-Zigha, A.; Aszodi, J.; Le Beller, D.; Süssmuth, R.D. The anti-staphylococcal lipolanthines are ribosomally synthesized lipopeptides. Nat. Chem. Biol. 2018, 14, 652–654. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
| Cpd. | R1 | R2 | Growth Inhibition (%) (a) | FAR Value (a) |
|---|---|---|---|---|
| 4 | 2-Cl | H | 9 | 11.20 |
| 5 | 2-Me | H | 34 | 8.19 |
| 6 | 2-Cl | 3-OMe | 37 | 32.62 |
| 7 | 2-Me | 3-OMe | 40 | 33.59 |
| 8 | 2-Cl | 4-OMe | 24 | 52.49 |
| 9 | 2-Me | 4-OMe | 2 | 30.80 |
| 10 | 2-Cl | 3-, 4-OMe | 9 | 59.76 |
| 11 | 2-Me | 3-, 4-OMe | n.a. (b) | 68.30 |
| 12 | 2-Cl | 3-OBn | n.a. (b) | 31.46 |
| 13 | 2-Me | 3-OBn | 17 | 42.29 |
| 14 | 4-OMe | H | 60 | 3.9 |
| 15 | 4-OMe | 3-OMe | 41 | 3.4 |
| 16 | 4-OMe | 4-OMe | 35 | 1.2 |
| 17 | 4-OMe | 3-, 4-OMe | 59 | 5.2 |
| 18 | 4-OMe | 3-OBn | 7 | n.d. (c) |
| INH | 97 | n.d. (c) | ||
| Verapamil | n.d. (c) | 4.10 | ||
| Tariquidar | n.d. (c) | 15.3 |
| Growth (%) | Growth (%) (a) | Percentual Growth | |
|---|---|---|---|
| Cpd. | Target Compound Only | Clofazimine + Target Compound Inhibition Increase (%) | |
| 4 | 96 | 28 | 44 |
| 11 | 100 | 23 | 55 |
| 12 | 100 | 34 | 44 |
| 13 | 81 | 28 | 54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lentz, F.; Reiling, N.; Spengler, G.; Kincses, A.; Csonka, A.; Molnár, J.; Hilgeroth, A. Dually Acting Nonclassical 1,4-Dihydropyridines Promote the Anti-Tuberculosis (Tb) Activities of Clofazimine. Molecules 2019, 24, 2873. https://doi.org/10.3390/molecules24162873
Lentz F, Reiling N, Spengler G, Kincses A, Csonka A, Molnár J, Hilgeroth A. Dually Acting Nonclassical 1,4-Dihydropyridines Promote the Anti-Tuberculosis (Tb) Activities of Clofazimine. Molecules. 2019; 24(16):2873. https://doi.org/10.3390/molecules24162873
Chicago/Turabian StyleLentz, Fabian, Norbert Reiling, Gabriella Spengler, Annamária Kincses, Andrea Csonka, Joseph Molnár, and Andreas Hilgeroth. 2019. "Dually Acting Nonclassical 1,4-Dihydropyridines Promote the Anti-Tuberculosis (Tb) Activities of Clofazimine" Molecules 24, no. 16: 2873. https://doi.org/10.3390/molecules24162873
APA StyleLentz, F., Reiling, N., Spengler, G., Kincses, A., Csonka, A., Molnár, J., & Hilgeroth, A. (2019). Dually Acting Nonclassical 1,4-Dihydropyridines Promote the Anti-Tuberculosis (Tb) Activities of Clofazimine. Molecules, 24(16), 2873. https://doi.org/10.3390/molecules24162873