
Synthesis of a Naphthalocyanine-Like Dye: The First Report on Zn(II)-1,6-methano[10]annulenecyanine



Abstract
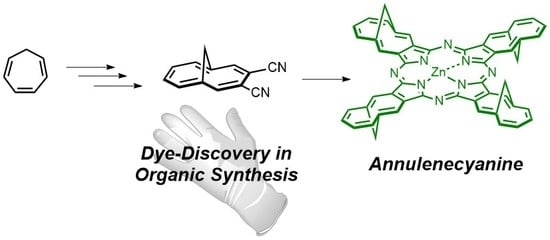
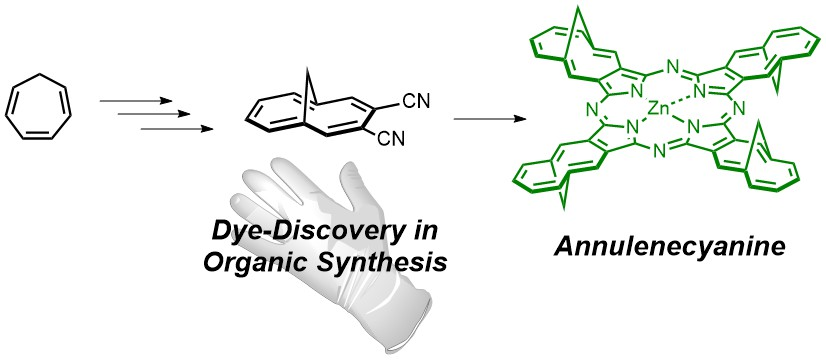
:
{kind=link}
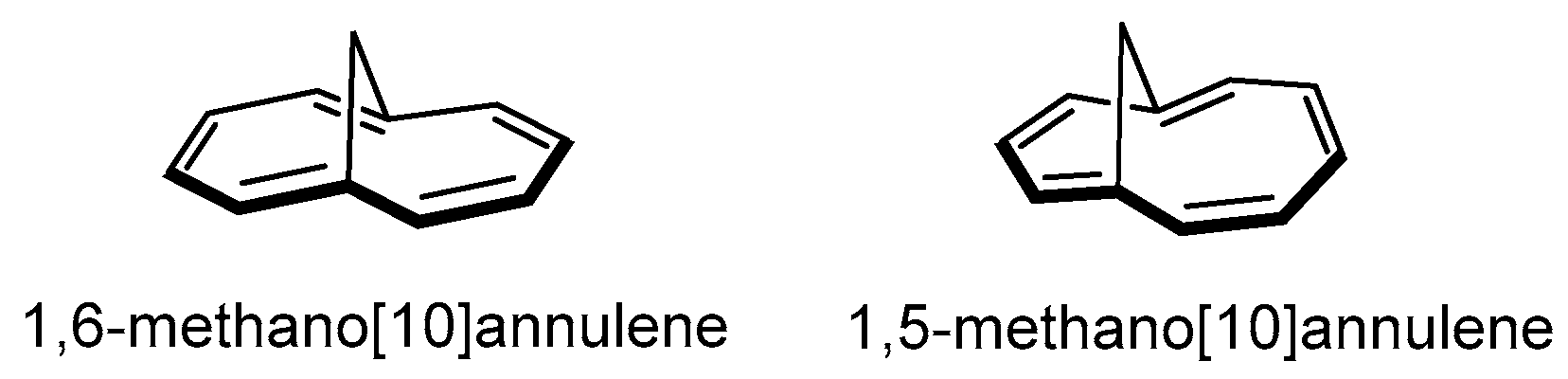{kind=link}
{kind=link}
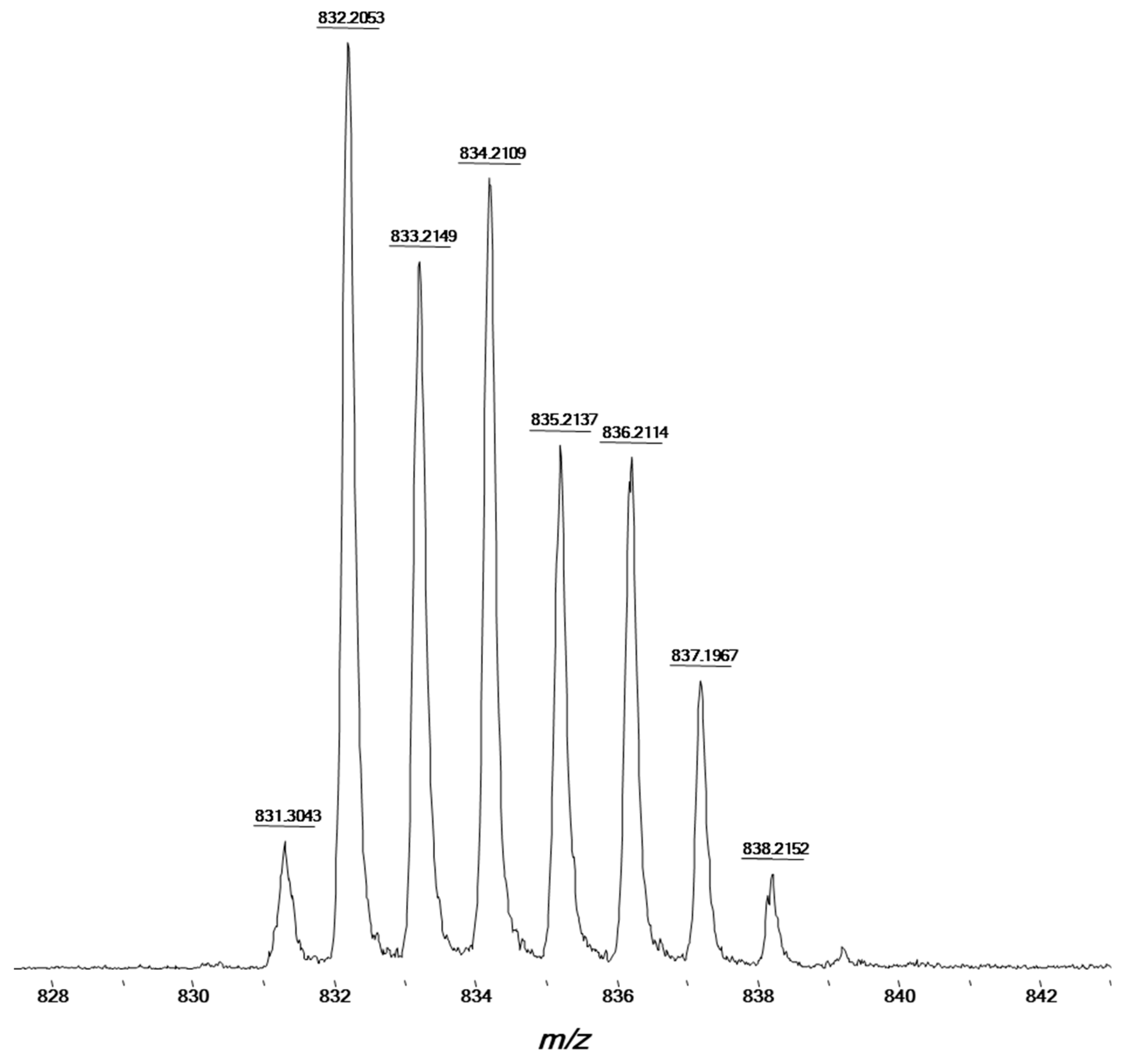{kind=link}
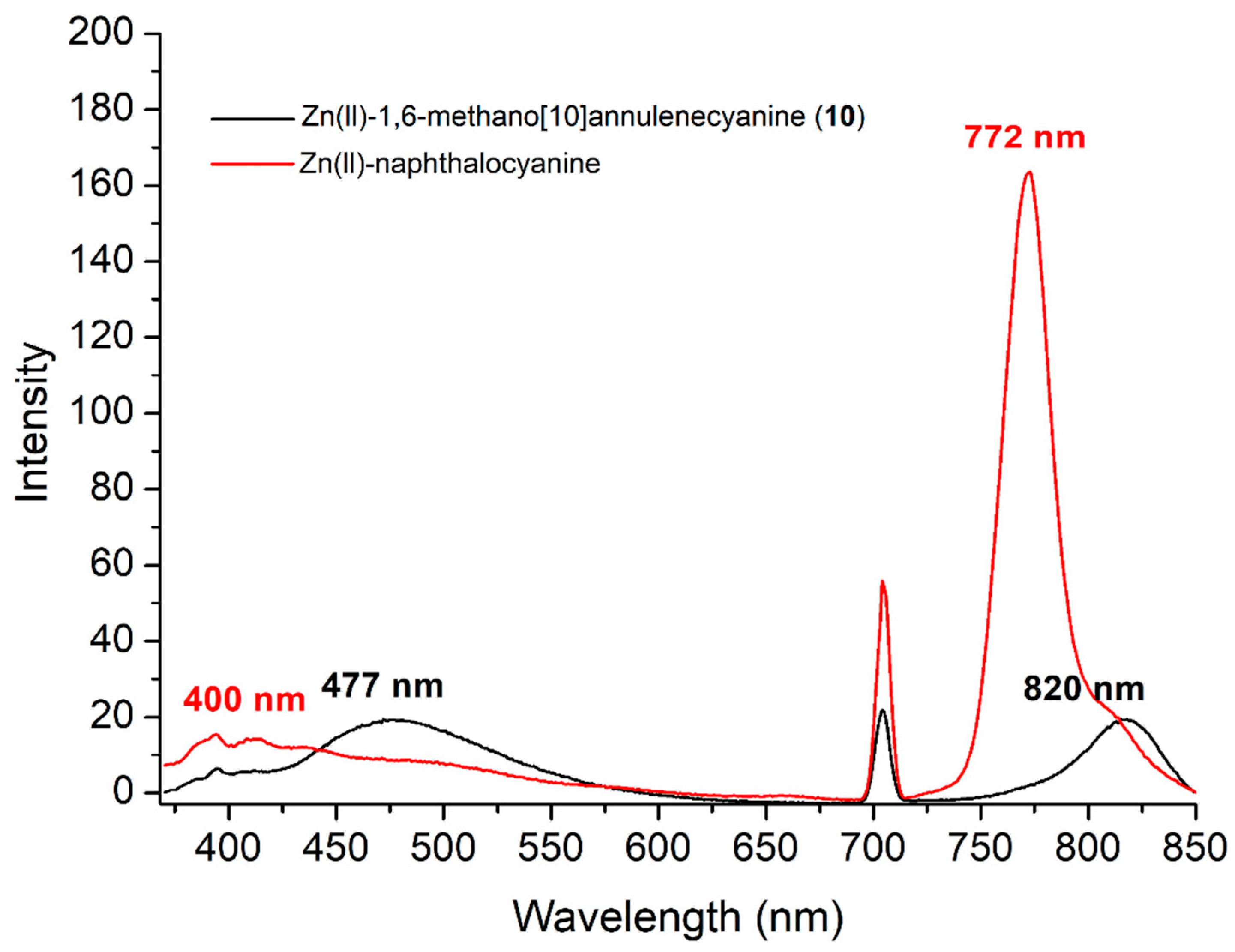{kind=link}
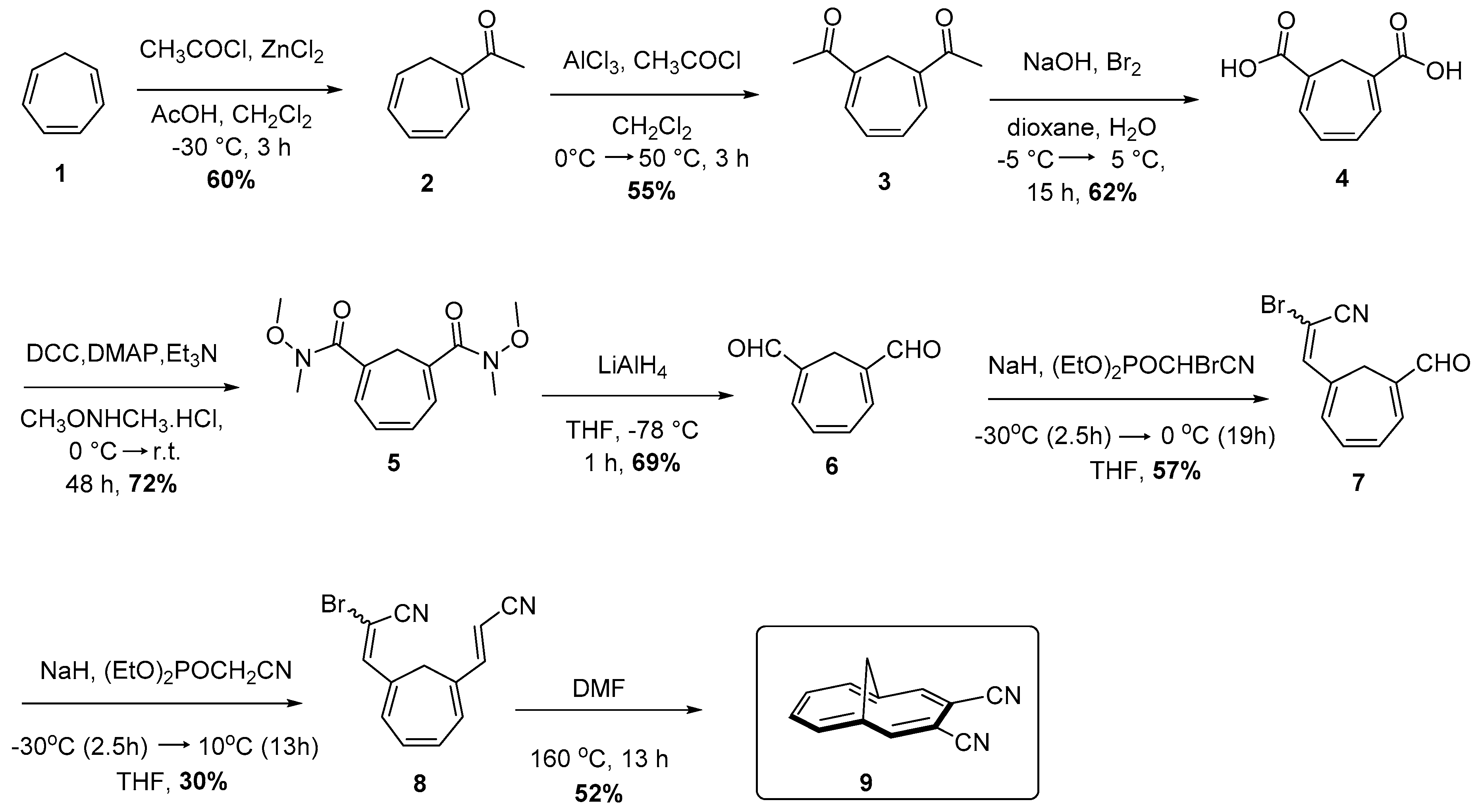{kind=link}
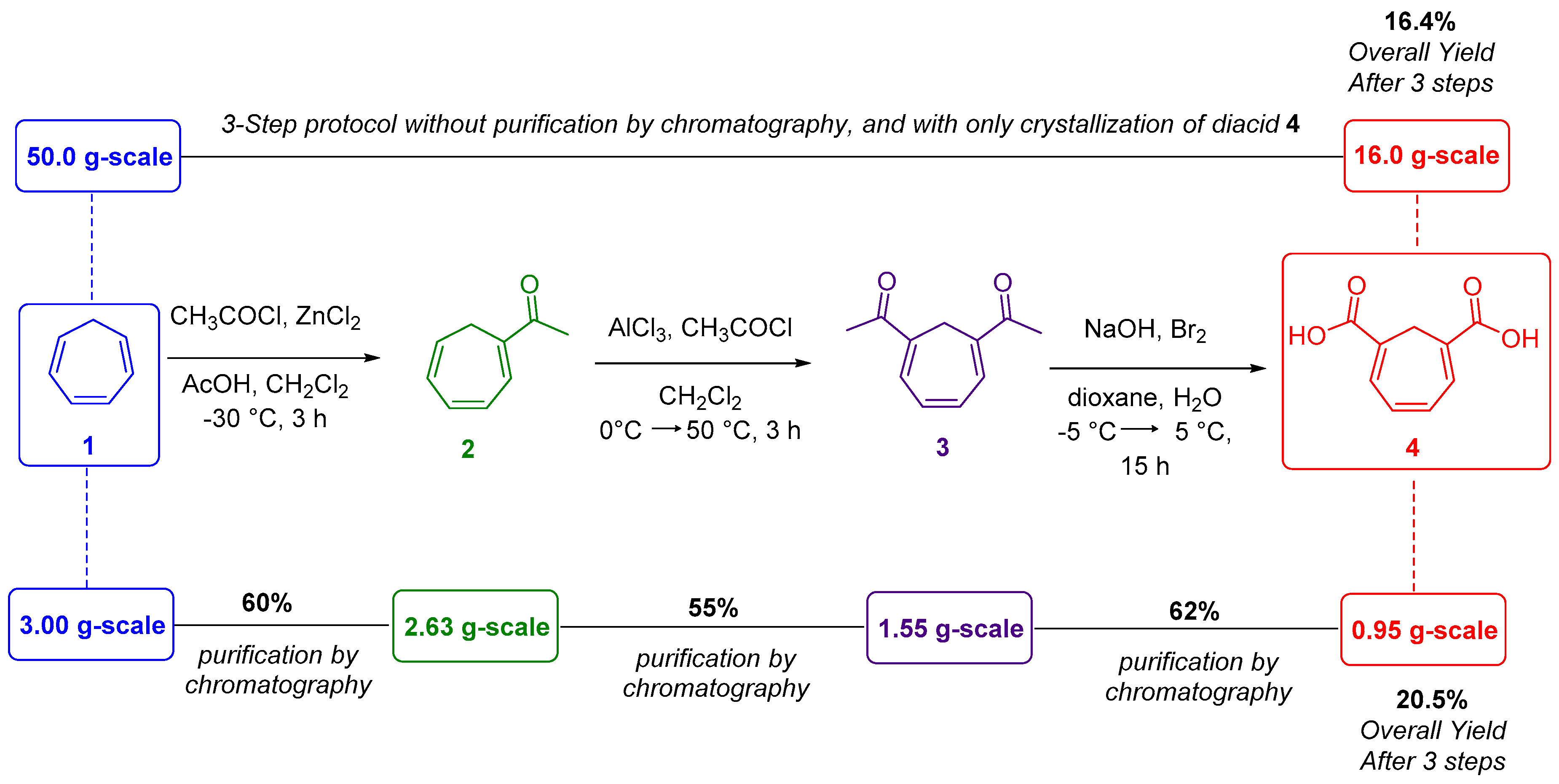{kind=link}
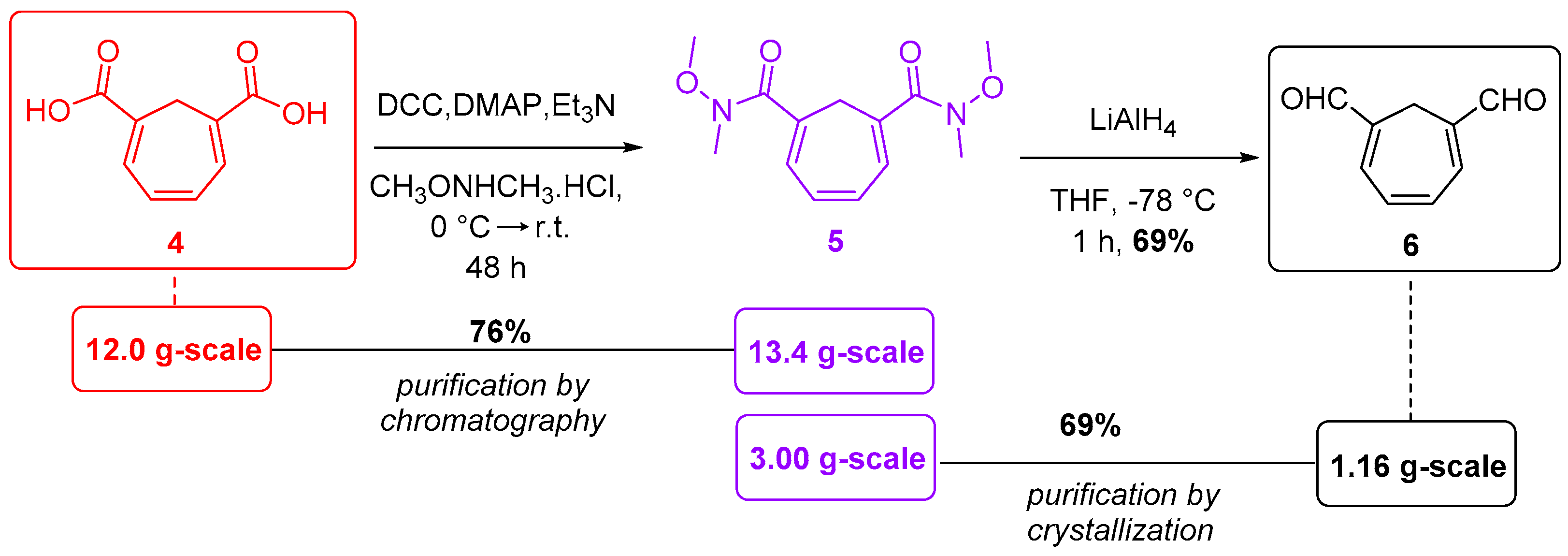{kind=link}
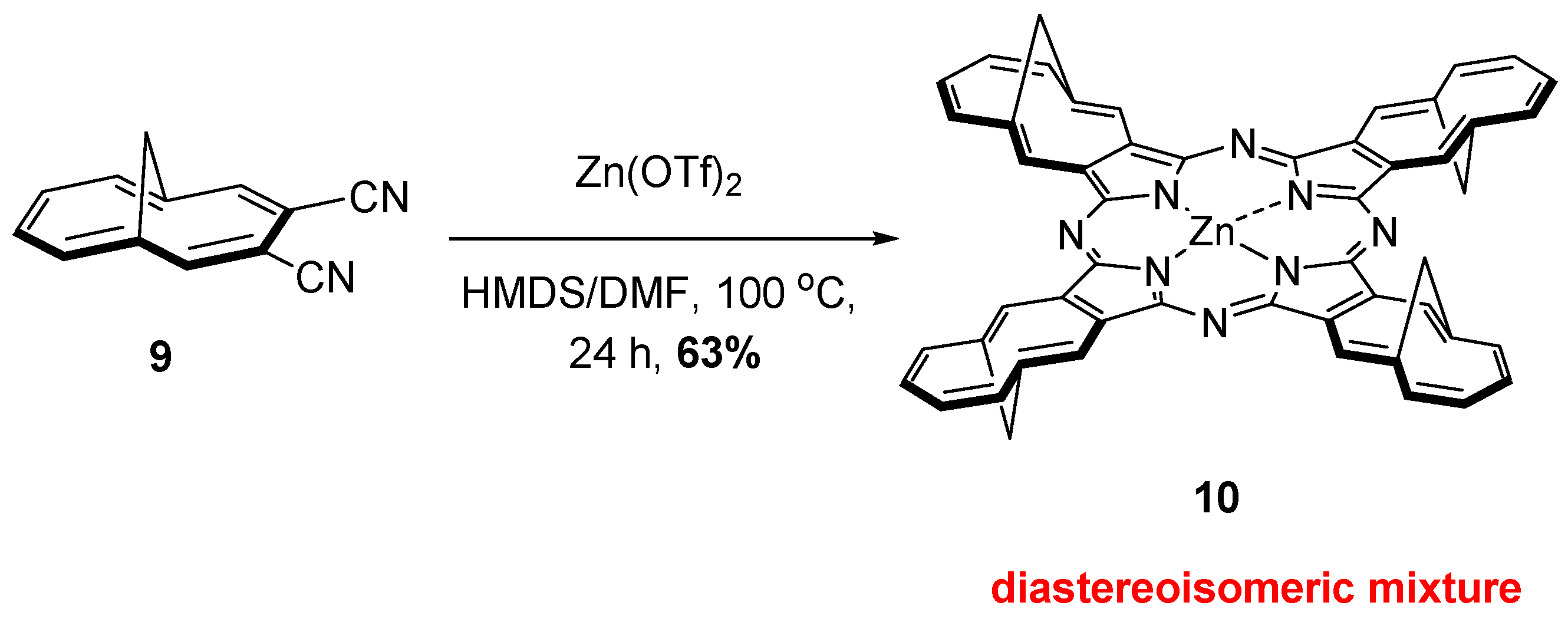{kind=link}
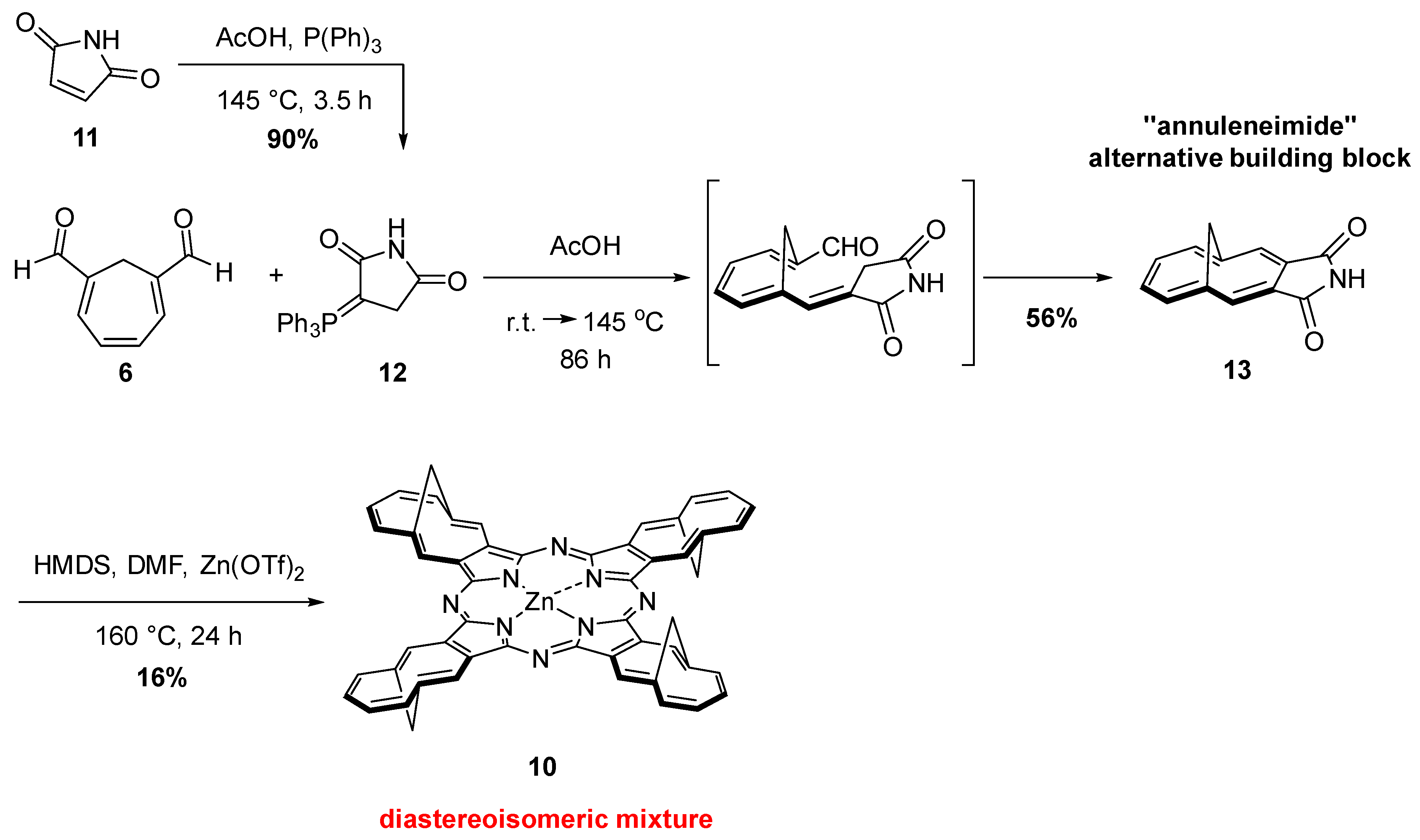{kind=link}
1. Introduction
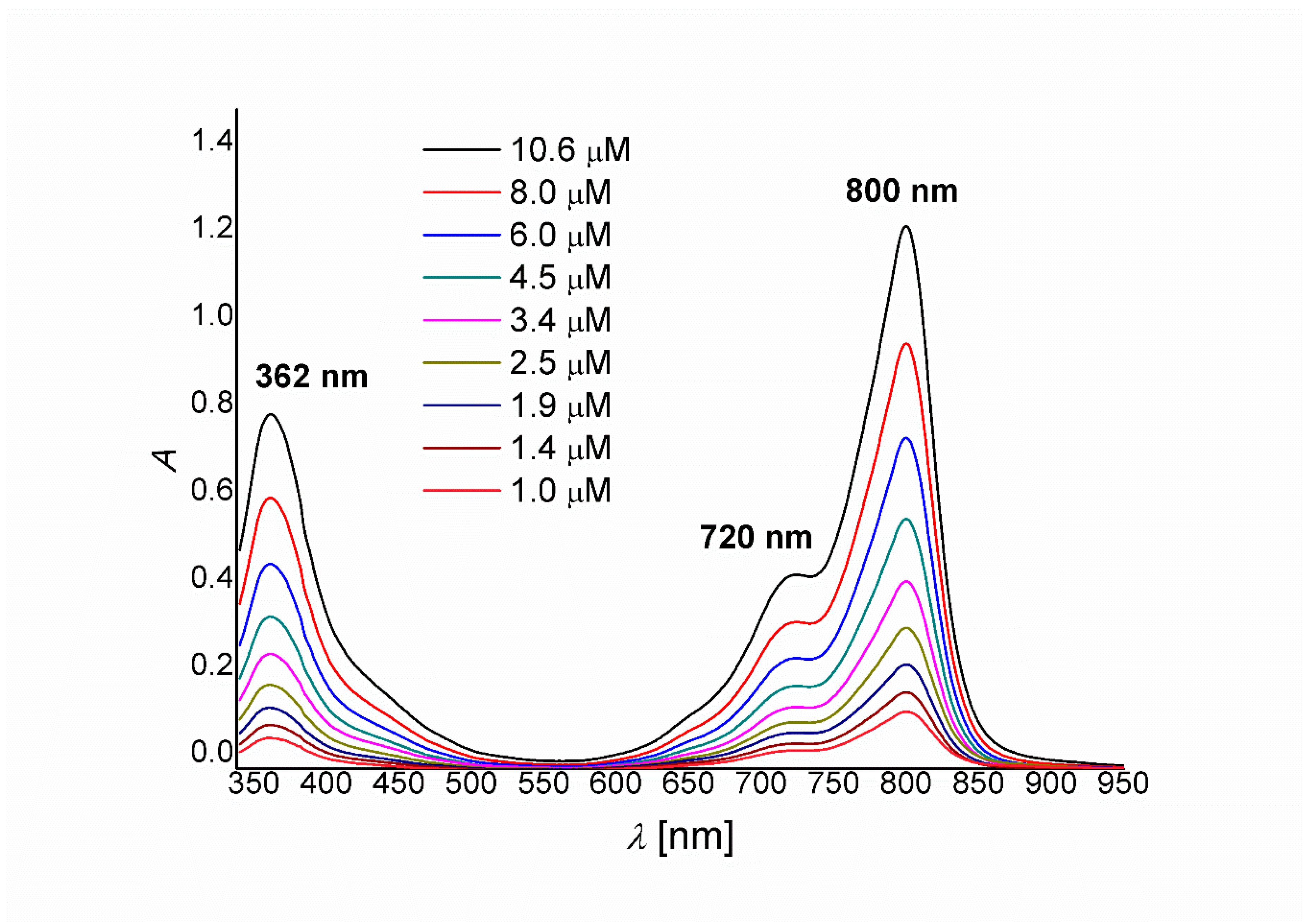
2. Results and Discussion
Synthesis
3. Materials and Methods
4. Experimental Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Urbani, M.; de la Torre, G.; Nazeeruddin, M.K.; Torres, T. Phthalocyanines and porphyrinoid analogues as hole- and electron-transporting materials for perovskite solar cells. Chem. Soc. Rev. 2019, 48, 2738–2766. [Google Scholar] [CrossRef]
- Molina, D.; Guerrero, A.; Garcia-Belmonte, G.; Fernández-Lázaro, F.; Sastre-Santos, Á. Synthesis of a Fully Conjugated Phthalocyanine-Diketopyrrolopyrrole-Phthalocyanine Triad as Low Band Gap Donor in Small Molecule Bulk Heterojunction Solar Cells. Eur. J. Org. Chem. 2014, 2014, 4585–4591. [Google Scholar] [CrossRef] [Green Version]
- Kuprikova, N.M.; Klyamer, D.D.; Sukhikh, A.S.; Krasnov, P.O.; Mrsic, I.; Basova, T.V. Fluorosubstituted lead phthalocyanines: Crystal structure, spectral and sensing properties. Dyes Pigments 2020, 173, 107939. [Google Scholar] [CrossRef]
- Aliabad, H.A.R.; Bashi, M. Cobalt phthalocyanine polymer for optoelectronic and thermoelectric applications. J. Mater. Sci. Mater. Electron. 2019, 30, 18720–18728. [Google Scholar] [CrossRef]
- Bechtold, I.H.; Eccher, J.; Faria, G.C.; Gallardo, H.; Molin, F.; Gobo, N.R.S.; de Oliveira, K.T.; von Seggern, H. New Columnar Zn-Phthalocyanine Designed for Electronic Applications. J. Phys. Chem. B 2012, 116, 13554–13560. [Google Scholar] [CrossRef] [PubMed]
- Pavani, C.; Francisco, C.M.L.; Gobo, N.R.S.; de Oliveira, K.T.; Baptista, M.S. Improved photodynamic activity of a dual phthalocyanine–ALA photosensitiser. New J. Chem. 2016, 40, 9666–9671. [Google Scholar] [CrossRef]
- Lourenço, L.M.O.; Rocha, D.M.G.C.; Ramos, C.I.V.; Gomes, M.C.; Almeida, A.; Faustino, M.A.F.; Almeida Paz, F.A.; Neves, M.G.P.M.S.; Cunha, Â.; Tomé, J.P.C. Photoinactivation of Planktonic and Biofilm Forms of Escherichia coli through the Action of Cationic Zinc(II) Phthalocyanines. ChemPhotoChem 2019, 3, 251–260. [Google Scholar] [CrossRef]
- Romero, M.P.; Gobo, N.R.S.; de Oliveira, K.T.; Iamamoto, Y.; Serra, O.A.; Louro, S.R.W. Photophysical properties and photodynamic activity of a novel menthol–zinc phthalocyanine conjugate incorporated in micelles. J. Photochem. Photobiol. A Chem. 2013, 253, 22–29. [Google Scholar] [CrossRef]
- Oshiro-Junior, J.A.; Sato, M.R.; Boni, F.I.; Santos, K.L.M.; de Oliveira, K.T.; de Freitas, L.M.; Fontana, C.R.; Nicholas, D.; McHale, A.; Callan, J.F.; et al. Phthalocyanine-loaded nanostructured lipid carriers functionalized with folic acid for photodynamic therapy. Mater. Sci. Eng. C 2020, 108, 110462. [Google Scholar] [CrossRef]
- Negri, L.B.; Martins, T.J.; da Silva Gobo, N.R.; de Oliveira, K.T.; Hamblin, M.R.; da Silva, R.S. Design, synthesis and photobiological activity of novel ruthenium phthalocyanine complexes. Inorg. Chem. Commun. 2019, 99, 60–63. [Google Scholar] [CrossRef]
- Revuelta-Maza, M.Á.; Mascaraque, M.; González-Jiménez, P.; González-Camuñas, A.; Nonell, S.; Juarranz, Á.; de la Torre, G.; Torres, T. Assessing Amphiphilic ABAB Zn(II) Phthalocyanines with Enhanced Photosensitization Abilities in In Vitro Photodynamic Therapy Studies Against Cancer. Molecules 2020, 25, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, A.C.S.; Silva, A.D.; Tomé, V.A.; Pinto, S.M.A.; Silva, E.F.F.; Calvete, M.J.F.; Gomes, C.M.F.; Pereira, M.M.; Arnaut, L.G. Phthalocyanine Labels for Near-Infrared Fluorescence Imaging of Solid Tumors. J. Med. Chem. 2016, 59, 4688–4696. [Google Scholar] [CrossRef]
- Lo, P.-C.; Rodríguez-Morgade, M.S.; Pandey, R.K.; Ng, D.K.P.; Torres, T.; Dumoulin, F. The unique features and promises of phthalocyanines as advanced photosensitisers for photodynamic therapy of cancer. Chem. Soc. Rev. 2020, 49, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento, F.B.; Manieri, T.M.; Cerchiaro, G.; Ribeiro, A.O. Synthesis of unsymmetrical phthalocyanine derivatives and their interaction with mammary MCF7 cells. Dyes Pigments 2013, 99, 316–322. [Google Scholar] [CrossRef]
- De Assis, F.F.; Ferreira, M.A.B.; Brocksom, T.J.; de Oliveira, K.T. NIR bacteriochlorin chromophores accessed by Heck and Sonogashira cross-coupling reactions on a tetrabromobacteriochlorin derivative. Org. Biomol. Chem. 2016, 14, 1402–1412. [Google Scholar] [CrossRef]
- Barona-Castaño, J.; Carmona-Vargas, C.; Brocksom, T.; de Oliveira, K. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulié, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Işci, U.; Berlinguette, C.P.; et al. CO2 electrochemical catalytic reduction with a highly active cobalt phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef]
- De Souza, T.; Antonio, F.; Zanotto, M.; Homem-de-Mello, P.; Ribeiro, A. Photophysical and Photochemical Properties and Aggregation Behavior of Phthalocyanine and Naphthalocyanine Derivatives. J. Braz. Chem. Soc. 2017, 29, 1199–1209. [Google Scholar] [CrossRef]
- De A. Bartolomeu, A.; Brocksom, T.J.; da Silva Filho, L.C.; de Oliveira, K.T. Multicomponent reactions mediated by NbCl 5 for the synthesis of phthalonitrile-quinoline dyads: Methodology, scope, mechanistic insights and applications in phthalocyanine synthesis. Dyes Pigments 2018, 151, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Pekbelgin Karaoğlu, H.; Kalkan Burat, A. α- and β-Substituted Metal-Free Phthalocyanines: Synthesis, Photophysical and Electrochemical Properties. Molecules 2020, 25, 363. [Google Scholar] [CrossRef] [Green Version]
- Abid, S.; Ben Hassine, S.; Richy, N.; Camerel, F.; Jamoussi, B.; Blanchard-Desce, M.; Mongin, O.; Paul, F.; Paul-Roth, C.O. Phthalocyanine-Cored Fluorophores with Fluorene-Containing Peripheral Two-Photon Antennae as Photosensitizers for Singlet Oxygen Generation. Molecules 2020, 25, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, T.F.M.; Ribeiro, A.O. The impact of the extended π-conjugation in photophysical, photochemical and aggregation behavior of new phthalocyanine–naphthalocyanine hybrids. J. Photochem. Photobiol. A Chem. 2017, 340, 1–7. [Google Scholar] [CrossRef]
- Gobo, N.R.S.; Brocksom, T.J.; Zukerman-Schpector, J.; De Oliveira, K.T. Synthesis of an octa-tert-butylphthalocyanine: A low-aggregating and photochemically stable photosensitizer. Eur. J. Org. Chem. 2013, 5028–5031. [Google Scholar] [CrossRef]
- Oliveira, K.; Momo, P.; Assis, F.; Ferreira, M.; Brocksom, T. Chlorins: Natural Sources, Synthetic Developments and Main Applications. Curr. Org. Synth. 2014, 11, 42–58. [Google Scholar] [CrossRef]
- De Oliveira, K.T.; de Souza, J.M.; Gobo, N.R.d.S.; de Assis, F.F.; Brocksom, T.J. Basic Concepts and Applications of Porphyrins, Chlorins and Phthalocyanines as Photosensitizers in Photonic Therapies. Rev. Virtual Quím. 2015, 7, 310–335. [Google Scholar]
- Gobo, N.R.S.; Brocksom, T.J.; de Oliveira, K.T. Soluble and Non-Aggregated Phthalocyanines: Synthesis, Mechanistic Aspects and Their Main Building Blocks. Curr. Org. Synth. 2018, 14, 1132–1155. [Google Scholar] [CrossRef]
- Sekkat, N.; Van Den Bergh, H.; Nyokong, T.; Lange, N. Like a bolt from the blue: Phthalocyanines in biomedical optics. Molecules 2012, 17, 98–144. [Google Scholar] [CrossRef] [Green Version]
- Lukyanets, E.A.; Nemykin, V.N. The key role of peripheral substituents in the chemistry of phthalocyanines and their analogs. J. Porphyr. Phthalocyanines 2010, 14, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Long, L.; Zhang, C. Synthesis of unsymmetrical phthalocyanines: A brief overview. Tetrahedron 2012, 68, 2433–2451. [Google Scholar] [CrossRef]
- Dumoulin, F.; Durmuş, M.; Ahsen, V.; Nyokong, T. Synthetic pathways to water-soluble phthalocyanines and close analogs. Coord. Chem. Rev. 2010, 254, 2792–2847. [Google Scholar] [CrossRef]
- Mack, J.; Kobayashi, N. Low Symmetry Phthalocyanines and Their Analogues. Chem. Rev. 2011, 111, 281–321. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.F.; de Oliveira, K.T.; Galembeck, S.E.; Bultinck, P.; Constantino, M.G. Aromaticity and Homoaromaticity in Methano[10]annulenes. J. Org. Chem. 2007, 72, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Vogel, E.; Roth, H.D. Synthese eines Cyclodecapentaens. Angew. Chem. 1964, 76, 145. [Google Scholar] [CrossRef]
- Barrett, D.G.; Gellman, S.H. Effects of amphiphile topology on aggregation properties: Distinctive behavior of contrafacial amphiphiles. J. Am. Chem. Soc. 1993, 115, 9343–9344. [Google Scholar] [CrossRef]
- Vogel, E.; Schäfer-Ridder, M.; Wagner, A. Neue Cycloheptatrien-Verbindungen und Verfahren zur Herstellung von Cycloheptatrien-Verbindungen. EP0011669A1, 30 November 1978. [Google Scholar]
- Sarmah, C.S.; Kataky, J.C.S. A novel synthesis of substituted 1,6- methano[10]annulenes using a phase transfer catalyst. Indian J. Chem. Sect. B Org. Chem. Incl. Med. Chem. 1993, 32B, 1149–1150. [Google Scholar]
- Uchida, H.; Reddy, P.Y.; Nakamura, S.; Toru, T. Novel Efficient Preparative Method for Phthalocyanines from Phthalimides and Phthalic Anhydride with HMDS. J. Org. Chem. 2003, 68, 8736–8738. [Google Scholar] [CrossRef]
- Oda, M.; Nakamura, T.; Neha, M.; Miyawaki, D.; Ohta, A.; Kuroda, S.; Miyatake, R. A Short-Step Synthesis of 1,6-Methano[10]annulene-3,4-dicarboximides and Their Benzene-, Naphthalene-, and Thiophene-Annulated Compounds. Eur. J. Org. Chem. 2014, 2014, 5976–5985. [Google Scholar] [CrossRef]
- Kuroda, S.; Oda, M.; Tamura, N.; Miyatake, R.; Matsumoto, N.; Horino, Y.; Miyawaki, D. Synthesis and Emission Properties of 1,6-Methano[10]annulene-3,4-dicarboximides. Heterocycles 2011, 83, 789. [Google Scholar] [CrossRef]
- Ogunsipe, A.; Chen, J.; Nyokong, T. Photophysical and photochemical studies of zinc(ii) phthalocyanine derivatives—effects of substituents and solvents. New J. Chem. 2004, 28, 822–827. [Google Scholar] [CrossRef]
- Wheeler, B.L.; Nagasubramanian, G.; Bard, A.J.; Schechtman, L.A.; Kenney, M.E. A silicon phthalocyanine and a silicon naphthalocyanine: Synthesis, electrochemistry, and electrogenerated chemiluminescence. J. Am. Chem. Soc. 1984, 106, 7404–7410. [Google Scholar] [CrossRef]
Sample availability: Samples not available. |









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gobo, N.R.d.S.; Brocksom, T.J.; de Oliveira, K.T. Synthesis of a Naphthalocyanine-Like Dye: The First Report on Zn(II)-1,6-methano[10]annulenecyanine. Molecules 2020, 25, 2164. https://doi.org/10.3390/molecules25092164
Gobo NRdS, Brocksom TJ, de Oliveira KT. Synthesis of a Naphthalocyanine-Like Dye: The First Report on Zn(II)-1,6-methano[10]annulenecyanine. Molecules. 2020; 25(9):2164. https://doi.org/10.3390/molecules25092164
Chicago/Turabian StyleGobo, Nicholas Roberto da Silva, Timothy John Brocksom, and Kleber Thiago de Oliveira. 2020. "Synthesis of a Naphthalocyanine-Like Dye: The First Report on Zn(II)-1,6-methano[10]annulenecyanine" Molecules 25, no. 9: 2164. https://doi.org/10.3390/molecules25092164
APA StyleGobo, N. R. d. S., Brocksom, T. J., & de Oliveira, K. T. (2020). Synthesis of a Naphthalocyanine-Like Dye: The First Report on Zn(II)-1,6-methano[10]annulenecyanine. Molecules, 25(9), 2164. https://doi.org/10.3390/molecules25092164