
Intracluster Sulphur Dioxide Oxidation by Sodium Chlorite Anions: A Mass Spectrometric Study


Abstract
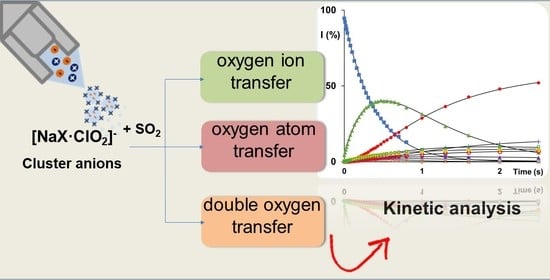
:
1. Introduction
2. Results and Discussion
2.1. Reactivity of [Cl·Na·ClO2]− Cluster Anion
Effect of the Ligand
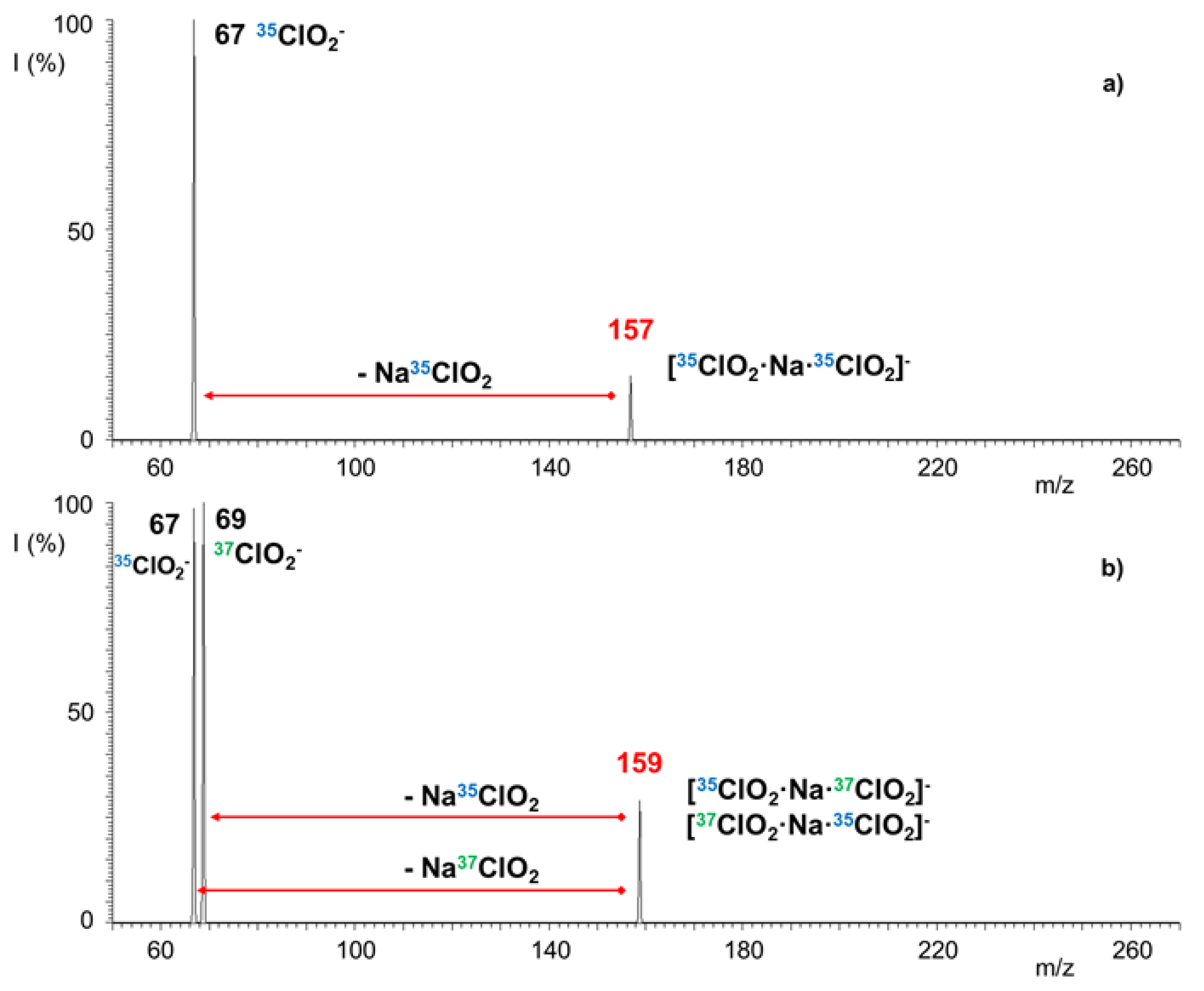
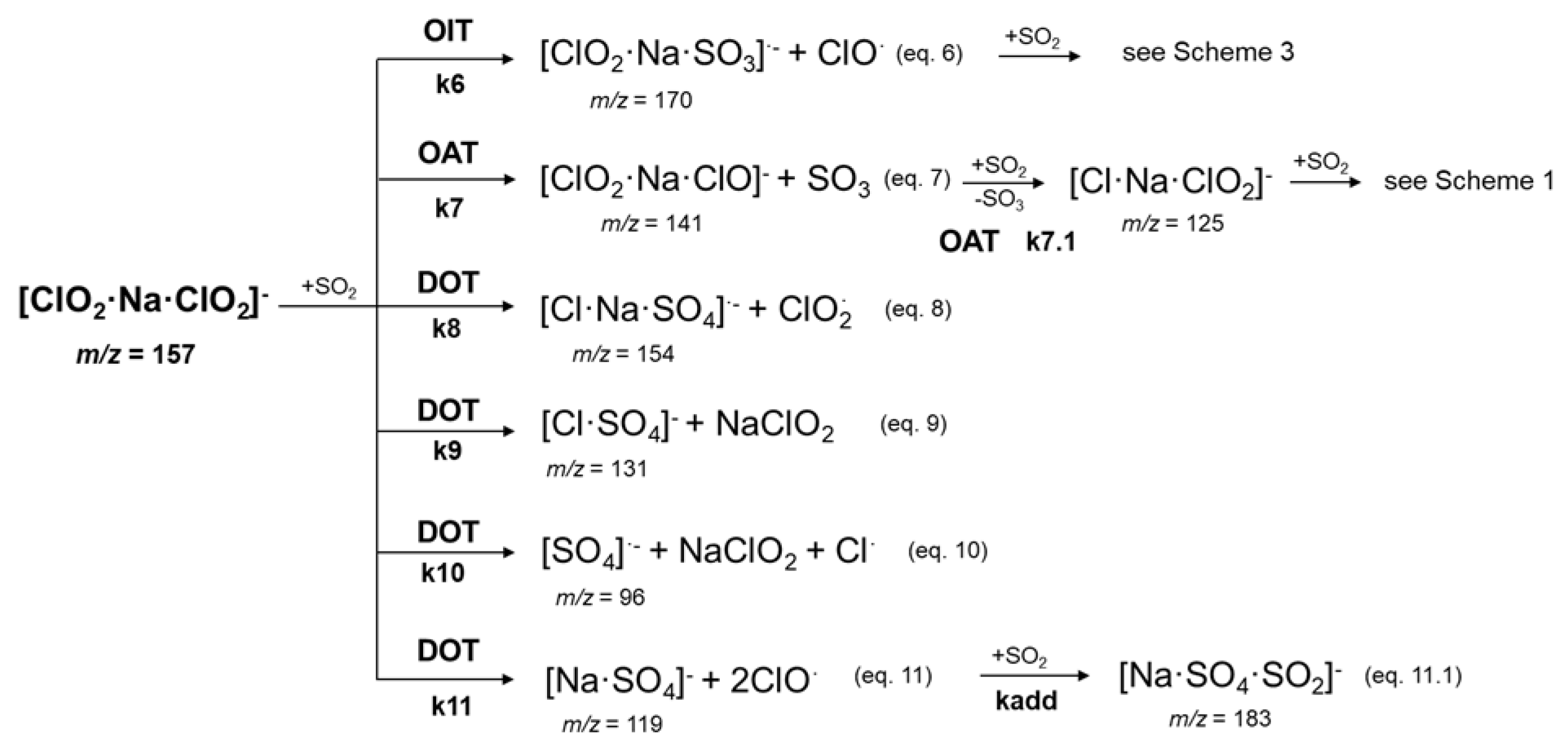
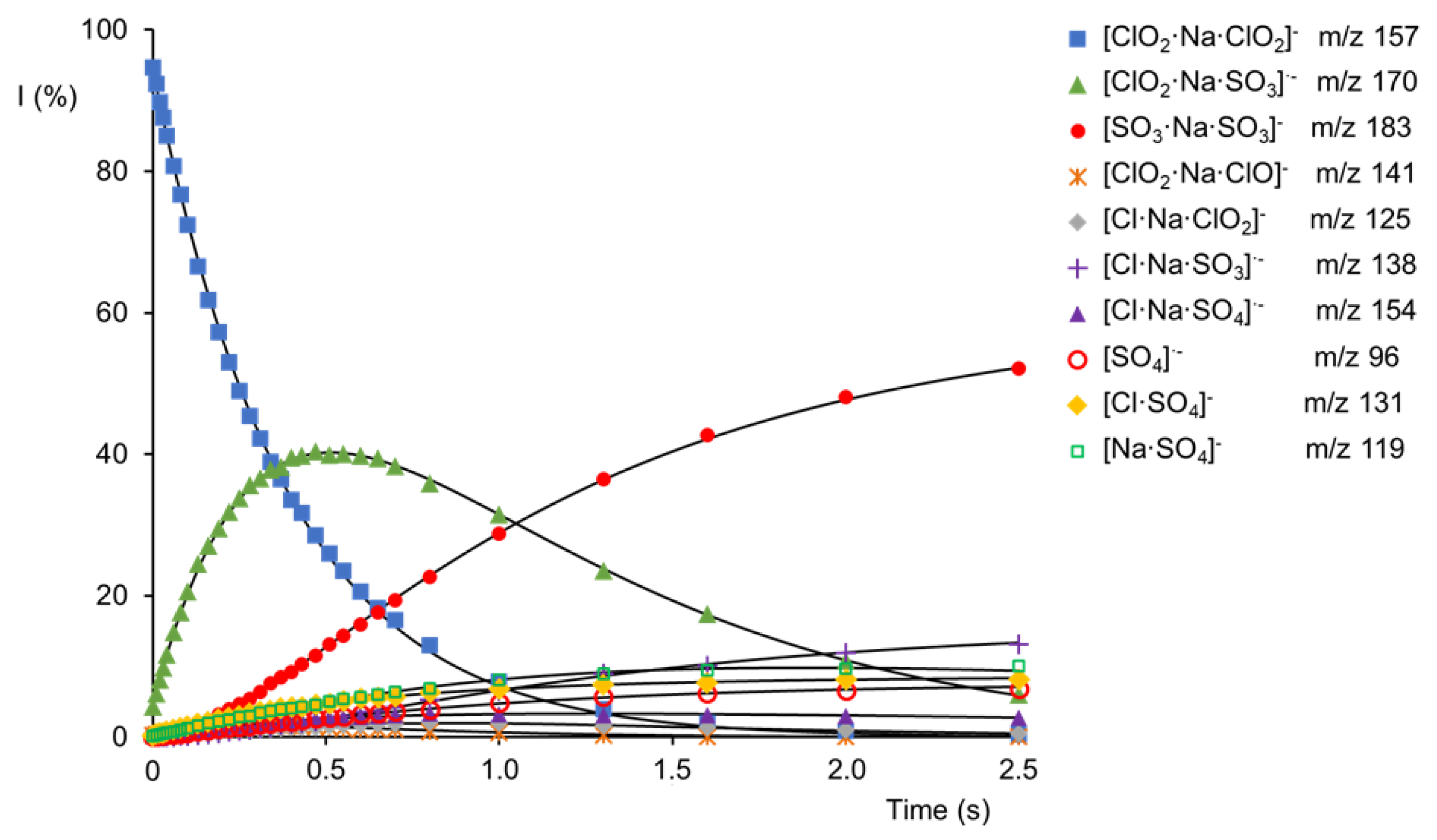
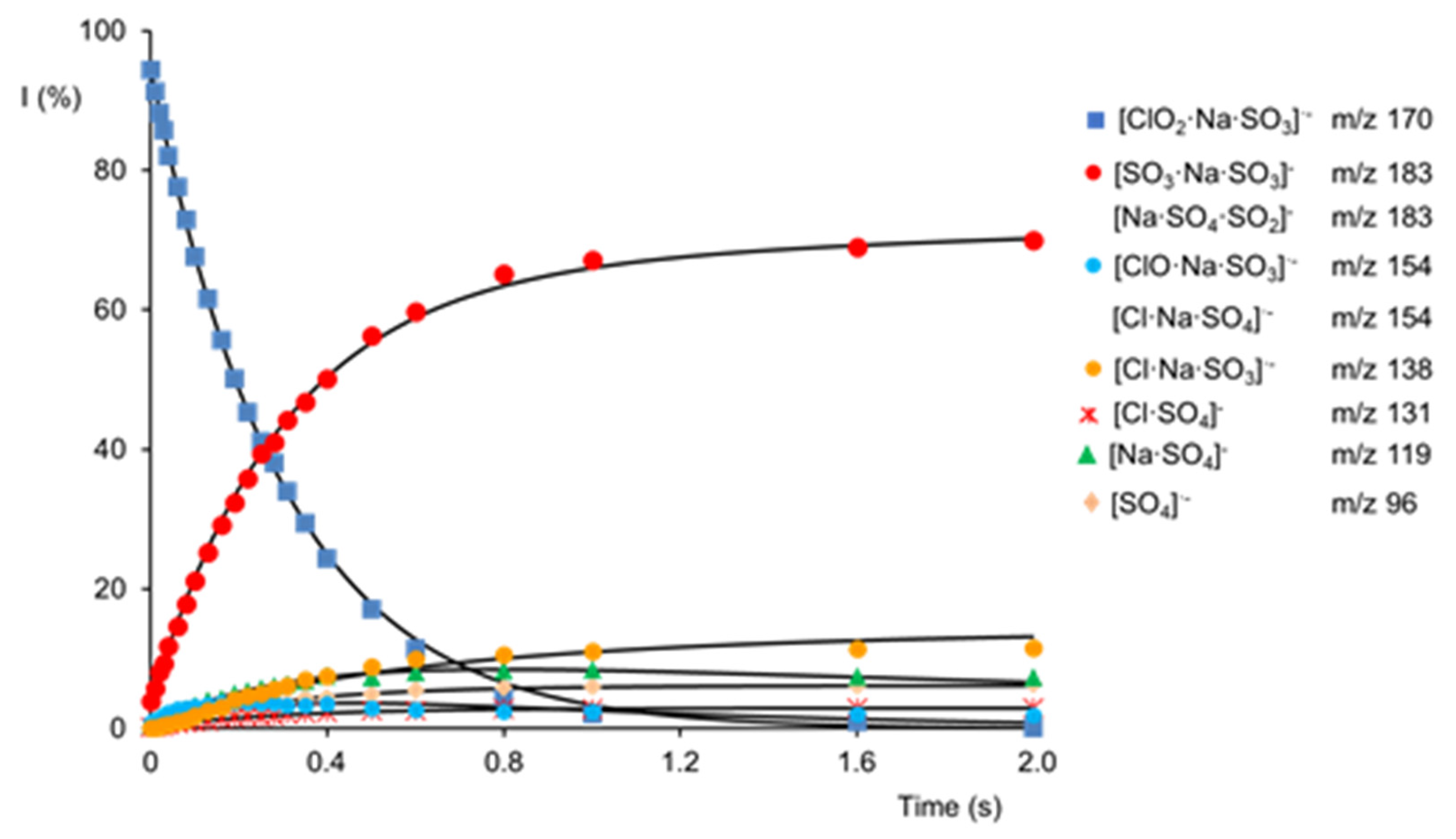
2.2. Reactivity of [ClO2·Na·ClO2]− Cluster Anion
2.3. Reactivity of [SO3·Na·ClO2]·− Cluster Anion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lima, R.; Bachmann, R.T. Pollutant emissions from modern incinerators. Int. J. Environ. Pollut. 2002, 18, 336. [Google Scholar] [CrossRef]
- Vehlow, J. Air pollution control systems in WtE units: An overview. Waste Manag. 2015, 37, 58–74. [Google Scholar] [CrossRef]
- Muralikrishna, I.V.; Manickam, V. Environmental Management; Elsevier Inc.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3rd ed.; John Wiley and Sons: New York, NY, USA, 2016. [Google Scholar]
- Dupart, Y.; King, S.M.; Nekat, B.; Nowak, A.; Wiedensohler, A.; Herrmann, H.; David, G.; Thomas, B.; Miffre, A.; Rairoux, P.; et al. Mineral dust photochemistry induces nucleation events in the presence of SO2. Proc. Natl. Acad. Sci. USA 2012, 109, 20842–20847. [Google Scholar] [CrossRef] [Green Version]
- Directive (EU) 2016/2284 of the European Parliament and of the Council of 14 December 2016 on the Reduction of National Emissions of Certain Atmospheric Pollutants, Amending Directive 2003/35/EC and Repealing Directive 2001/81/EC.
- Woodard & Curran, Inc. Industrial Waste Treatment Handbook, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Hu, G.; Dam-Johansen, K.; Wedel, S.; Peterhansen, J. Review of the direct sulfation reaction of limestone. Prog. Energy Combust. Sci. 2006, 32, 386–407. [Google Scholar] [CrossRef]
- Koralegedara, N.H.; Pinto, P.X.; Dionysiou, D.; Al-Abed, S.R. Recent advances in flue gas desulphurization gypsum processes and applications—A review. J. Environ. Manag. 2019, 251, 109572. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, T.; Chen, Z. Experimental study on simultaneous desulfurization and denitrification from flue gas with composite absorbent. Environ. Prog. Sustain. Energy 2010, 30, 216–220. [Google Scholar] [CrossRef]
- Adewuyi, Y.G.; He, X.; Shaw, H.; Lolertpihop, W. Lolertpihop, Simultaneous absorption and oxidation of NO and SO2 by aqueous solutions of sodium chlorite. Chem. Eng. Commun. 1999, 174, 21–51. [Google Scholar] [CrossRef]
- Flagiello, D.; Di Natale, F.; Erto, A.; Lancia, A. Wet oxidation scrubbing (WOS) for flue-gas desulphurization using sodium chlorite seawater solutions. Fuel 2020, 277, 118055. [Google Scholar] [CrossRef]
- Chu, H.; Chien, T.-W.; Twu, B.-W. The absorption kinetics of NO in NaClO2/NaOH solutions. J. Hazard. Mater. 2001, B84, 241–252. [Google Scholar] [CrossRef]
- Chu, H.; Chien, T.-W.; Twu, B.-W. Simultaneous absorption of SO2 and no in a stirred tank reactor with NaClO2/NaOH solutions. Water Air Soil Pollut. 2003, 143, 337–350. [Google Scholar] [CrossRef]
- Pourmohammadbagher, A.; Jamshidi, E.; Ale-Ebrahim, H.; Dabir, S. Study on simultaneous removal of NOx and SO2 with NaClO2 in a novel swirl wet system. Ind. Eng. Chem. Res. 2011, 50, 8278–8284. [Google Scholar] [CrossRef]
- Chien, T.-W.; Chu, H. Removal of SO2 and NO from flue gas by wet scrubbing using an aqueous NaClO2 solution. J. Hazard. Mater. 2000, 80, 43–57. [Google Scholar] [CrossRef]
- Krzyżyńka, R.; Zhao, Y.; Hutson, N. Absorption of NOx, SO2, and mercury in a simulated additive-enhanced wet flue gas desulphurization scrubber. Polish J. Environ. Stud. 2010, 19, 1255–1262. [Google Scholar]
- Andreasen, A.; Mayer, S. Use of Seawater Scrubbing for SO2 Removal from Marine Engine Exhaust Gas. Energy Fuels 2007, 21, 3274–3279. [Google Scholar] [CrossRef]
- Byun, Y.; Hamilton, I.P.; Tu, X.; Shin, D.N. Formation of chlorinated species through reaction of SO2 with NaClO2 powder and their role in the oxidation of NO and Hg0. Environ. Sci. Pollut. Res. 2014, 21, 8052–8058. [Google Scholar] [CrossRef]
- Coelho, F.; Eberlin, M.N. The bridge connecting gas-phase and solution chemistries. Angew. Chem. Int. Ed. 2011, 50, 5261–5263. [Google Scholar] [CrossRef]
- Zhu, W.; Yuan, Y.; Zhou, P.; Zeng, L.; Wang, H.; Tang, L.; Guo, B.; Chen, B. The expanding role of electrospray ionization mass spectrometry for probing reactive intermediates in solution. Molecules 2012, 17, 11507–11537. [Google Scholar] [CrossRef]
- Fabris, D. Mass spectrometric approaches for the investigation of dynamic processes in condensed phase. Mass Spectrom. Rev. 2004, 24, 30–54. [Google Scholar] [CrossRef]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Iron-Promoted C-C Bond Formation in the Gas Phase. Angew. Chem. Int. Ed. 2015, 54, 14359–14362. [Google Scholar] [CrossRef]
- Troiani, A.; De Petris, G.; Pepi, F.; Garzoli, S.; Salvitti, C.; Rosi, M.; Ricci, A. Base-Assisted Conversion of Protonated D -Fructose to 5-HMF: Searching for Gas-Phase Green Models. ChemistryOpen 2019, 8, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Vikse, K.L.; Ahmadi, Z.; Manning, C.C.; Harrington, D.A.; McIndoe, J.S. Powerful insight into catalytic mechanisms through simultaneous monitoring of reactants, products, and intermediates. Angew. Chem. Int. Ed. 2011, 50, 8304–8306. [Google Scholar] [CrossRef] [PubMed]
- Gronert, S. Mass spectrometric studies of organic ion/molecule reactions. Chem. Rev. 2001, 101, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, I.; Frenich, A.G.; Romero-González, R. Mass spectrometry approaches to ensure food safety. Anal. Methods 2020, 12, 1148–1162. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Cai, Z. The latest developments and applications of mass spectrometry in food-safety and quality analysis. Trends Anal. Chem. 2013, 52, 170–185. [Google Scholar] [CrossRef]
- Cimino, P.; Troiani, A.; Pepi, F.; Garzoli, S.; Salvitti, C.; Di Rienzo, B.; Barone, V.; Ricci, A. From ascorbic acid to furan derivatives: The gas phase acid catalyzed degradation of vitamin C. Phys. Chem. Chem. Phys. 2018, 20, 17132–17140. [Google Scholar] [CrossRef]
- Geoghegan, K.F.; Kelly, M.A. Biochemical applications of mass spectrometry in pharmaceutical drug discovery. Mass Spectrom. Rev. 2005, 24, 347–366. [Google Scholar] [CrossRef]
- Loos, G.; Van Schepdael, A.; Cabooter, D. Quantitative mass spectrometry methods for pharmaceutical analysis. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150366. [Google Scholar] [CrossRef] [Green Version]
- Forte, G.; Chiarotto, I.; Giannicchi, I.; Loreto, M.A.; Martinelli, A.; Micci, R.; Pepi, F.; Rossi, S.; Salvitti, C.; Stringaro, A.; et al. Characterization of naproxen–polymer conjugates for drug-delivery. J. Biomater. Sci. Polym. Ed. 2015, 27, 69–85. [Google Scholar] [CrossRef]
- Vestal, M.L. The Future of Biological Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Finehout, E.J.; Lee, K.H. An introduction to mass spectrometry applications in biological research. Biochem. Mol. Biol. Educ. 2004, 32, 93–100. [Google Scholar] [CrossRef]
- de Laeter, J.R. Mass spectrometry and geochronology. Mass Spectrom. Rev. 1998, 17, 97–125. [Google Scholar] [CrossRef]
- Petrie, S.; Bohme, D.K. Ions in space. Mass Spectrom. Rev. 2006, 26, 258–280. [Google Scholar] [CrossRef] [PubMed]
- Cacace, F.; Cipollini, R.; DE Petris, G.; Troiani, A. The impervious route to the elusive HOOO− anion. Int. J. Mass Spectrom. 2003, 228, 717–722. [Google Scholar] [CrossRef]
- Cacace, F.; de Petris, G.; Pepi, F.; Troiani, A. Direct experimental evidence for the H2O+O2− charge transfer complex: Crucial support to atmospheric photonucleation theory. Angew. Chem. Int. Ed. 2000, 39, 367–369. [Google Scholar] [CrossRef]
- Cacace, F.; de Petris, G.; Pepi, F.; Rosi, M.; Troiani, A. Ionization of ozone/chlorofluorocarbon mixtures in atmospheric gases: Formation and remarkable dissociation of [CHXYO3]+ complexes (X = H, Cl, F; Y= Cl, F). Chem. Eur. J. 2000, 6, 2572–2581. [Google Scholar] [CrossRef]
- Johnson, G.; Tyo, E.C.; Castleman, A.W. Cluster reactivity experiments: Employing mass spectrometry to investigate the molecular level details of catalytic oxidation reactions. Proc. Natl. Acad. Sci. USA 2008, 105, 18108–18113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, G.E.; Mitrić, R.; Bonačić-Koutecký, V.; Castleman, A. Clusters as model systems for investigating nanoscale oxidation catalysis. Chem. Phys. Lett. 2009, 475, 1–9. [Google Scholar] [CrossRef]
- Schlangen, M.; Schwarz, H. Effects of ligands, cluster size, and charge state in gas-phase catalysis: A happy marriage of experimental and computational studies. Catal. Lett. 2012, 142, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Dietl, D.-C.N.; Troiani, A.; Schlangen, M.; Ursini, O.; Angelini, G.; Apeloig, Y.; de Petris, G.; Schwarz, H. Mechanistic Aspects of Gas-Phase Hydrogen-Atom Transfer from Methane to [CO].+ and [SiO].+: Why Do They Differ? Chem. Eur. J. 2013, 19, 6662–6669. [Google Scholar] [CrossRef]
- Schlangen, M.; Schwarz, H. Probing elementary steps of nickel-mediated bond activation in gas-phase reactions: Ligand- and cluster-size effects. J. Catal. 2011, 284, 126–137. [Google Scholar] [CrossRef]
- Vikse, K.L.; McIndoe, J.S. Mechanistic insights from mass spectrometry: Examination of the elementary steps of catalytic reactions in the gas phase. Pure Appl. Chem. 2015, 87, 361–377. [Google Scholar] [CrossRef] [Green Version]
- Vikse, K.L.; Ahmadi, Z.; McIndoe, J.S. The application of electrospray ionization mass spectrometry to homogeneous catalysis. Coord. Chem. Rev. 2014, 279, 96–114. [Google Scholar] [CrossRef]
- Troiani, A.; Rosi, M.; Salvitti, C.; de Petris, G. The oxidation of sulfur dioxide by single and double oxygen transfer paths. ChemPhysChem 2014, 15, 2723–2731. [Google Scholar] [CrossRef]
- Stewart, I.I. Electrospray mass spectrometry: A tool for elemental speciation. Spectrochim. Acta Part B 1999, 54, 1649–1695. [Google Scholar] [CrossRef]
- Hao, C.; March, R.E. Electrospray ionization tandem mass spectrometric study of salt cluster ions: Part 2 Salts of polyatomic acid groups and of multivalent metals. J. Mass Spectrom. 2001, 36, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Eberlin, M.N. Electrospray ionization mass spectrometry: A major tool to investigate reaction mechanisms in both solutions and the gas phase. Eur. J. Mass Spectrom. 2007, 13, 19–28. [Google Scholar] [CrossRef]
- Castleman, A.W., Jr.; Keesee, R.G. Gas-phase clusters: Spanning the states of matter. Science 1988, 241, 36–42. [Google Scholar] [CrossRef]
- Bondybey, V.E.; Beyer, M.K. How many molecules make a solution? Int. Rev. Phys. Chem. 2002, 21, 277–306. [Google Scholar] [CrossRef]
- Troiani, A.; Salvitti, C.; de Petris, G. Gas-phase reactivity of carbonate ions with sulphur dioxide: An experimental study of clusters reactions. J. Am. Soc. Mass Spectrom. 2019, 30, 1964–1972. [Google Scholar] [CrossRef]
- Shaik, S.; Mandal, D.; Ramanan, S.S.D.M.R. Oriented electric fields as future smart reagents in chemistry. Nat. Chem. 2016, 8, 1091–1098. [Google Scholar] [CrossRef]
- Yue, L.; Li, J.; Zhou, S.; Sun, X.; Schlangen, M.; Shaik, S.; Schwarz, H. Control of product distribution and mechanism by ligation electric field in the thermal activation of methane. Angew. Chem. Int. Ed. 2017, 56, 10219–10223. [Google Scholar] [CrossRef]
- Geng, C.; Li, J.; Schlangen, M.; Shaik, S.; Sun, X.; Wang, N.; Weiske, T.; Yue, L.; Zhou, S.; Schwarz, H. Oriented external electric fields as mimics for probing the role of metal ions and ligands in the thermal gas-phase activation of methane. Dalton Trans. 2018, 47, 15271–15277. [Google Scholar] [CrossRef]
- de Petris, G.; Cartoni, A.; Troiani, A.; Angelini, G.; Ursini, O. Ursini, Water activation by SO2+ ions: An effective source of OH radicals. PhysChemChemPhys 2009, 11, 9976–9978. [Google Scholar] [CrossRef]
- de Petris, G.; Troiani, A.; Rosi, M.; Angelini, G.; Ursini, O. Selective activation of C-Cl and C-F bonds by SO+ radical cations: An experimental and computational study. ChemPlusChem 2013, 78, 1065–1072. [Google Scholar] [CrossRef]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Vanadium hydroxide cluster ions in the gas phase: Bond-forming reactions of doubly-charged negative ions by SO2-promoted V-O activation. Chem. Eur. J. 2017, 23, 11752–11756. [Google Scholar] [CrossRef] [PubMed]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Sulphur dioxide cooperation in hydrolysis reactions of vanadium oxide and hydroxide cluster dianions. New J. Chem. 2018, 42, 4008–4016. [Google Scholar] [CrossRef]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Effective redox reactions by chromium oxide anions: Sulfur dioxide oxidation in the gas phase. Int. J. Mass Spectrom. 2018, 436, 18–22. [Google Scholar] [CrossRef]
- Salvitti, C.; Rosi, M.; Pepi, F.; Troiani, A.; de Petris, G. Reactivity of transition metal dioxide anions MO2− (M = Co, Ni, Cu, Zn) with sulfur dioxide in the gas phase: An experimental and theoretical study. Chem. Phys. Lett. 2021, 776, 138555. [Google Scholar] [CrossRef]
- E Bartmess, J.; Georgiadis, R. Empirical methods for determination of ionization gauge relative sensitivities for different gases. Vacuum 1983, 33, 149–153. [Google Scholar] [CrossRef]
- Kuzmic, P. Program DYNAFIT for the Analysis of Enzyme Kinetic Data: Application to HIV Proteinase. Anal. Biochem. 1996, 237, 260–273. [Google Scholar] [CrossRef]
- Bowers, M.T.; Su, T. Interactions between Ions and Molecules; Plenum Press: New York, NY, USA, 1975. [Google Scholar]
- Van Berkel, G.J.; Kertesz, V. Using the Electrochemistry of the Electrospray Ion Source. Anal. Chem. 2007, 79, 5510–5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linstrom, P.J.; Mallard, W.G. (Eds.) NIST Chemistry WebBook; NIST Standard Reference Database Number 69; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1997. Available online: http://webbook.nist.gov (accessed on 20 October 2021).
- Tro, N.J. Chemistry, International Edition; Pearson Education Inc.: Upper Saddle River, NJ, USA, 2016; pp. 341–343. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | kdec (×10−10) § | k/kcoll (%) | OIT | OAT | DOT |
|---|---|---|---|---|---|
| ClO2− + SO2 [47] | 9.10 | 63.8 | 86.5 | 4.2 | 9.3 |
| [Cl·Na·ClO2]− + SO2 | 2.88 | 24.2 | 74.2 | 8.8 | 17.0 |
| [F·Na·ClO2]− + SO2 | 3.75 | 30.8 | 89.2 | 3.7 | 7.1 |
| [Br·Na·ClO2]− + SO2 | 2.12 | 18.7 | 73.4 | 10.1 | 16.5 |
| [I·Na·ClO2]− + SO2 | 1.85 | 16.8 | 68.9 | 13.7 | 17.4 |
| [Cl·Li·ClO2]− + SO2 | 1.81 | 14.5 | 84.7 | 7.9 | 7.4 |
| [ClO2·Na·ClO2]− + SO2 | 7.48 | 66.0 | 81.8 | 4.8 | 13.4 |
| [SO3·Na·ClO2]− + SO2 | 3.74 | 33.0 | 70.4 | 6.9 | 22.7 |
| [(NaClO2)2·ClO2]− + SO2 | 7.39 | 68.1 | - | - | - |
| Reactivity | k × 10−10 cm3 mol−1 s−1 § | ||
|---|---|---|---|
| OIT | k1 | k2.2 | |
| 2.13 | 0.743 | ||
| OAT | k2 | k2.1 | |
| 0.251 | 7.53 | ||
| DOT | k3 | k4 | k5 |
| 0.306 | 0.147 | 0.034 | |
| Reactivity | k × 10−10 cm3 mol−1 s−1 § | |||
|---|---|---|---|---|
| OIT | k6 | |||
| 6.26 | ||||
| OAT | k7 | k7.1 | ||
| 0.364 | 9.57 | |||
| DOT | k8 | k9 | k10 | k11 |
| 0.251 | 0.417 | 0.134 | 0.222 | |
| SO2 add | kSO2 | |||
| 0.574 | ||||
| Reactivity | k × 10−10 cm3 mol−1 s−1 § | ||
|---|---|---|---|
| OIT | k12 | ||
| 2.63 | |||
| OAT | k13 | k13.1 | |
| 0.260 | 1.09 | ||
| DOT | k14 | k15 | k16 |
| 0.167 | 0.222 | 0.457 | |
| SO2 add | kadd | ||
| 0.494 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvitti, C.; Pepi, F.; Troiani, A.; de Petris, G. Intracluster Sulphur Dioxide Oxidation by Sodium Chlorite Anions: A Mass Spectrometric Study. Molecules 2021, 26, 7114. https://doi.org/10.3390/molecules26237114
Salvitti C, Pepi F, Troiani A, de Petris G. Intracluster Sulphur Dioxide Oxidation by Sodium Chlorite Anions: A Mass Spectrometric Study. Molecules. 2021; 26(23):7114. https://doi.org/10.3390/molecules26237114
Chicago/Turabian StyleSalvitti, Chiara, Federico Pepi, Anna Troiani, and Giulia de Petris. 2021. "Intracluster Sulphur Dioxide Oxidation by Sodium Chlorite Anions: A Mass Spectrometric Study" Molecules 26, no. 23: 7114. https://doi.org/10.3390/molecules26237114