
Modifications of Plasma Membrane Organization in Cancer Cells for Targeted Therapy

 , ,
, ,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. The Multidrug Resistance Phenomenon and Its Modulators
2.1. Types of Cancer Multidrug Resistance and Its Mechanisms
2.2. Proteins Determining MDR
2.3. MDR Modulators
Modulators of Natural Origin
3. MicroRNAs as Regulators of MDR Gene Expression
4. Anticancer Therapies Targeting Ion Channels
5. Plasma Membrane Receptors and Modulations
5.1. The Mechanism of Signal Transduction from The External Environment to The Cell by Integral Membrane Receptor
5.2. Reaction Required and Amplification Mechanism
5.3. Participation of Integral Membrane Receptors in the Pathological Metabolic Pathway and Targeted Therapy
6. Modulation of Cell Membrane Lipidomics
7. Electroporation
7.1. The Aqueous Pore Formation
7.2. Effects of The Electric Field on Oxidative Stress
7.3. Effects of the Electric Field on Lipid Peroxidation
7.4. Effects of Electric Pulses on Membrane Proteins
7.5. Cytoskeleton Remodeling after PEF
7.6. Clinical Potential (Aspect) of Electroporation
8. Sonoporation
8.1. In Vitro Research on Sonoporation
8.2. Preclinical Studies on Sonoporation
8.3. Clinical Application of Sonoporation in Oncology
9. Gravitational Forces Affecting Biomembranes
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Preta, G. New Insights into Targeting Membrane Lipids for Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 876. [Google Scholar] [CrossRef]
- Zalba, S.; Ten Hagen, T.L.M. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Szlasa, W.; Zendran, I.; Zalesińska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef]
- Kojima, K. Molecular aspects of the plasma membrane in tumor cells. Nagoya J. Med. Sci. 1993, 56, 1–18. [Google Scholar] [PubMed]
- Nicolson, G.L. Cell Membrane Fluid–Mosaic Structure and Cancer Metastasis. Cancer Res. 2015, 75, 1169–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivel, T.; Ramseyer, C.; Yesylevskyy, S. The asymmetry of plasma membranes and their cholesterol content influence the uptake of cisplatin. Sci. Rep. 2019, 9, 5627. [Google Scholar] [CrossRef] [Green Version]
- Zhen, X.; Cheng, P.; Pu, K. Recent Advances in Cell Membrane–Camouflaged Nanoparticles for Cancer Phototherapy. Small 2019, 15, e1804105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakhomova, O.N.; Gregory, B.W.; Pakhomov, A.G. Facilitation of electroporative drug uptake and cell killing by electrosensitization. J. Cell. Mol. Med. 2013, 17, 154–159. [Google Scholar] [CrossRef]
- Neumann, E.; Kakorin, S. Membrane electroporation: Chemical thermodynamics and flux kinetics revisited and refined. Eur. Biophys. J. 2018, 47, 373–387. [Google Scholar] [CrossRef]
- Lakshmanan, S.; Gupta, G.K.; Avci, P.; Chandran, R.; Sadasivam, M.; Jorge, A.E.S.; Hamblin, M.R. Physical energy for drug delivery; poration, concentration and activation. Adv. Drug Deliv. Rev. 2014, 71, 98–114. [Google Scholar] [CrossRef] [Green Version]
- Belyavskaya, N.A. Free and membrane-bound calcium in microgravity and microgravity effects at the membrane level. Adv. Sp. Res. Off. J. Comm. Sp. Res. 1996, 17, 169–177. [Google Scholar] [CrossRef]
- Kohn, F.; Hauslage, J.; Hanke, W. Membrane Fluidity Changes, A Basic Mechanism of Interaction of Gravity with Cells? Microgravity Sci. Technol. 2017, 29, 337–342. [Google Scholar] [CrossRef]
- Rojas, M.; Donahue, J.P.; Tan, Z.; Lin, Y.Z. Genetic engineering of proteins with cell membrane permeability. Nat. Biotechnol. 1998, 16, 370–375. [Google Scholar] [CrossRef]
- Yan, H.; Shao, D.; Lao, Y.-H.; Li, M.; Hu, H.; Leong, K.W. Engineering Cell Membrane-Based Nanotherapeutics to Target Inflammation. Adv. Sci. 2019, 6, 1900605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Fan, X.; Wu, Y.; Ma, Q.; Xu, Q.; Xie, X.; Chen, N. Mutation of genes for cell membrane synthesis in Corynebacterium glutamicum causes temperature-sensitive trait and promotes L-glutamate excretion. Biotechnol. Biotechnol. Equip. 2020, 34, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Minko, T.; Rodriguez-Rodriguez, T.; Pozharov, V. Nanotechnology approaches for personalized treatment of multidrug resistant cancer. Adv. Drug Deliv. Rev. 2013, 65, 1880–1895. [Google Scholar] [CrossRef] [Green Version]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Lehne, G. P-glycoprotein as a drug target in the treatment of multidrug resistant cancer. Curr. Drug Targets 2000, 1, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M. Expression of a multidrug resistance gene in human cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. 1), 35–48. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Heindl, L.M.; Bauman, P.A.; Ludi, C.W.; Dalton, W.S.; Cress, A.E. Cytokeratin expression results in a drug-resistant phenotype to six different chemotherapeutic agents. Clin. Cancer Res. 1996, 2, 97–105. [Google Scholar] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumor heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, A.J.; Jassem, J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs 2011, 71, 1385–1396. [Google Scholar] [CrossRef]
- Stavrovskaya, A.A.; Stromskaya, T.P. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry 2008, 73, 592–604. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. Mechanism of ABC transporters: A molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc. Natl. Acad. Sci. USA 2002, 99, 12639–12644. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.M.; Schindler, M. Cell biological mechanisms of multidrug resistance in tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 3497–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilers, M.; Roy, U.; Mondal, D. MRP (ABCC) transporters-mediated efflux of anti-HIV drugs, saquinavir and zidovudine, from human endothelial cells. Exp. Biol. Med. 2008, 233, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Bansal, T.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J. Pharm. Pharm. Sci. 2009, 12, 46–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, C.L.; Pulaski, L.; Gottesman, M.M.; Pastan, I. Analysis of the properties of the N-terminal nucleotide-binding domain of human P-glycoprotein. Biochemistry 2000, 39, 5518–5526. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kim, I.W.; Sauna, Z.E. The power of the pump: Mechanisms of action of P-glycoprotein (ABCB1). Eur. J. Pharm. Sci. 2006, 27, 392–400. [Google Scholar] [CrossRef]
- Tandon, V.R.; Kapoor, B.; Bano, G.; Gupta, S.; Gillani, Z.; Gupta, S.; Kour, D. P-glycoprotein: Pharmacological relevance. Indian J. Pharmacol. 2006, 38, 13–24. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Kamis, A.B.; Collaghan, R.; Higgins, C.F.; Ford, R.C. Three dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J. Biol. Chem. 2003, 278, 8294–8299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, T.W.; Clarke, D.M. Do drug substrates enter the common drug-binding pocket of P-glycoprotein through “gates”? Biochem. Biophys. Res. Commun. 2005, 329, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.M.; Hait, W.N. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol. Rev. 1990, 42, 155–199. [Google Scholar] [PubMed]
- Yang, K.; Wu, J.; Li, X. Recent advances in the research of P-glycoprotein inhibitors. Biosci. Trends 2008, 2, 137–146. [Google Scholar]
- Lampidis, T.J.; Krishan, A.; Planas, L.; Tapiero, H. Reversal of intrinsic resistance to adriamycin in normal cells by verapamil. Cancer Drug. Deliv. 1986, 3, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hollt, V.; Kouba, M.; Dietel, M.; Vogt, G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem. Pharmacol. 1992, 43, 2601–2608. [Google Scholar] [CrossRef]
- Popęda, M.; Płuciennik, E.; Bednarek, A.K. Proteins in cancer multidrug resistance. Postepy Hig. Med. Dosw. 2014, 68, 616–632. [Google Scholar] [CrossRef]
- Borowski, E.; Bontemps-Gracz, M.M.; Piwkowska, A. Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochim. Pol. 2005, 52, 609–627. [Google Scholar] [CrossRef]
- Liscovitch, M.; Lavie, Y. Cancer multidrug resistance: A review of recent drug discovery research. Drugs 2002, 5, 349–355. [Google Scholar]
- Perez-Tomas, R. Multidrug resistance: Retrospect and prospects in anti-cancer drug treatment. Curr. Med. Chem. 2006, 13, 1859–1876. [Google Scholar] [CrossRef]
- Robey, R.W.; Massey, P.R.; Amiri-Kordestani, L.; Bates, S.E. ABC transporters: Unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med. Chem. 2010, 10, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Lim, L.Y.; Chowbay, B. Herbal modulation of P-glycoprotein. Drug Metab. Rev. 2004, 36, 57–104. [Google Scholar] [CrossRef]
- Zhou, S.F.; Zhou, Z.W.; Li, C.G.; Chen, X.; Yu, X.; Xue, C.C.; Herington, A. Identification of drugs that interact with herbs in drug development. Drug Discov. Today 2007, 12, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, Y.; Wang, Q.; Chen, Z.; Li, X.; Wu, Z.; Hu, C.; Liao, D.; Zhang, W.; Chen, Z.S. The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol. Cancer 2020, 19, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Yang, X.; Morris, M.E. Flavonoids Are Inhibitors of Breast Cancer Resistance Protein (ABCG2)-Mediated Transport. Mol. Pharmacol. 2004, 65, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Ghadia, R.; Dand, N. BCS class IV drugs: Highly notorious candidates forformulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Smith, M.M.; Muller, M.; Kerr, K.M.; Ambudkar, S.V. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J. Bioenerg. Biomembr. 2001, 33, 481–491. [Google Scholar] [CrossRef]
- Regev, R.; Assaraf, Y.G.; Eytan, G.D. Membrane fluidization by ether, other anesthetics, and certain agents abolishes P-glycoprotein ATPase activity and modulates efflux from multidrug-resistant cells. Eur. J. Biochem. 1999, 259, 18–24. [Google Scholar] [CrossRef]
- Sachs-Barrable, K.; Thamboo, A.; Lee, S.D.; Wasan, K.M. Lipid excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 cells. J. Pharm. Pharm. Sci. 2007, 10, 319–331. [Google Scholar] [PubMed]
- Drori, S.; Eytan, G.D.; Assaraf, Y.G. Potentiation of anticancer-drug cytotoxicity by multidrug-resistance chemosensitizers involves alterations in membrane fluidity leading to increased membrane permeability. Eur. J. Biochem. 1995, 228, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Byrem, T.M.; Nair, M.G.; Strasburg, G.M. Modulation of liposomal membrane fluidity by flavonoids and isoflavonoids. Arch. Biochem. Biophys. 2000, 373, 102–109. [Google Scholar] [CrossRef]
- Boumendjel, A.; Di Pietro, A.; Dumontet, C.; Barron, D. Recent advances in the discovery of flavonoids and analogues with high affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med. Res. Rev. 2002, 22, 512–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Morris, M.E. Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-glycoprotein-mediated transport. J. Pharmacol. Exp. Ther. 2003, 304, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Conseil, G.; Baubichon-Cortay, H.; Dayan, G.; Jault, J.M.; Barron, D.; Di Pietro, A. Flavonoids: A class of modulators with bifunctional interactions at vicinal ATP- and steroid-binding sites on mouse P-glycoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 9831–9836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumendjel, A.; Beney, C.; Deka, N.; Mariotte, A.M.; Lawson, M.A.; Trompier, D.; Baubichon-Cortay, H.; Di Pietro, A. 4-Hydroxy-6-methoxyaurones with high-affinity binding to cytosolic domain of P-glycoprotein. Chem. Pharm. Bull. 2002, 50, 854–856. [Google Scholar] [CrossRef] [Green Version]
- Critchfield, J.W.; Welsh, C.J.; Phang, J.M.; Yeh, G.C. Modulation of adriamycin accumulation and efflux by flavonoids in HCT-15 colon cells: Activation of P-glycoprotein as a putative mechanism. Biochem. Pharmacol. 1994, 48, 1437–1445. [Google Scholar] [CrossRef]
- Phang, J.M.; Poore, C.M.; Lopaczynska, J.; Yeh, G.C. Flavonol stimulated efflux of 7,12-dimethylbenz(a)-anthracene in multidrug-resistant breast cancer cells. Cancer Res. 1993, 53, 5977–5981. [Google Scholar] [PubMed]
- Scambia, G.; Ranelletti, F.O.; Panici, P.B.; De Vincenzo, R.; Bonanno, G.; Ferrandina, G.; Piantelli, M.; Bussa, S.; Rumi, C.; Cianfriglia, M.; et al. Quercetin potentiates the effect of adriamycin in a multidrug-resistant MCF-7 human breast-cancer cell line: P-glycoprotein as a possible target. Cancer Chemother. Pharmacol. 1994, 34, 459–464. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ling, V. Effect of quercetin on Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem. Pharmacol. 1997, 53, 587–596. [Google Scholar] [CrossRef]
- Choi, J.S.; Shin, S.C. Enhanced paclitaxel bioavailability after oral coadministration of paclitaxel prodrug with naringin to rats. Int. J. Pharm. 2005, 292, 149–156. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Yang, Y.; Cai, C.Y.; Teng, Q.X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist. Updates 2021, 54, 100743. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Bootchai, C.; Ngamphiw, C.; Tongsima, S. microPIR: An integrated database of microRNA target sites within human promoter sequences. PLoS ONE 2012, 7, e33888. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, L.; Hu, J.; Ruan, J. miR-138 might reverse multidrug resistance of leukemia cells. Leuk. Res. 2010, 34, 1078–1082. [Google Scholar] [CrossRef]
- Zhang, H.; Li, M.; Han, Y.; Zheng, X. Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma. Dig. Dis. Sci. 2010, 55, 2545–2551. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Rodrigues, V.; Seca, H.; Sousa, D.; Sousa, E.; Lima, R.T.; Vasconcelos, M.H. The network of P-glycoprotein and microRNAs interactions. Int. J. Cancer 2014, 135, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, O.; Filkowski, J.; Meservy, J.; Ilnytskyy, Y.; Tryndyak, V.P.; Chekhun, V.F.; Pogribny, I.P. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol. Cancer Ther. 2008, 7, 2152–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.D.; Zhang, H.; Zhang, P.; Zheng, Y.S.; Zhang, X.J.; Han, B.W.; Luo, X.Q.; Xu, L.; Zhou, H.; Qu, L.H.; et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J. Cell Mol. Med. 2011, 15, 2164–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Hazari, S.; Mehra, S.; Kaushal, D.; Moroz, K.; Dash, S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am. J. Pathol. 2012, 180, 2490–2503. [Google Scholar] [CrossRef] [Green Version]
- Ikemura, K.; Yamamoto, M.; Miyazaki, S.; Mizutani, H.; Iwamoto, T.; Okuda, M. MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells. Mol. Pharmacol. 2013, 83, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hu, S.; Wang, J.; Cai, J.; Xiao, L.; Yu, L.; Wang, Z. MiR-27a modulates MDR1/P-glycoprotein expression by targeting HIPK2 in human ovarian cancer cells. Gynecol. Oncol. 2010, 119, 125–130. [Google Scholar] [CrossRef]
- Shang, Y.; Feng, B.; Zhou, L.; Ren, G.; Zhang, Z.; Fan, X.; Sun, Y.; Luo, G.; Liang, J.; Wu, K.; et al. The miR27b-CCNG1-P53-miR-508-5p axis regulates multidrug resistance of gastric cancer. Oncotarget 2016, 7, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.; Spevak, C.C.; Wong, G.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction with protein kinase C(epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the Production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J. Biol. Chem. 2009, 284, 26533–26546. [Google Scholar] [PubMed] [Green Version]
- Haenisch, S.; Werk, A.N.; Cascorbi, I. MicroRNAs and their relevance to ABC transporters. Br. J. Clin. Pharmacol. 2014, 77, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Yang, L.; Hu, J. Down-regulation of miR-27a might inhibit proliferation and drug resistance of gastric cancer cells. J. Exp. Clin. Cancer Res. 2011, 30, 55. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, J.; Wang, J.; Gao, W.; Ding, Y.; Ding, Y.; Jia, Z. Down-regulation of miR-210-3p encourages chemotherapy resistance of renal cell carcinoma via modulating ABCC1. Cell Biosci. 2018, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.Z.; Morris, M.E.; Yu, A.M. MicroRNA-328 negatively regulates the expression of breast cancer resistance protein (BCRP/ABCG2) in human cancer cells. Mol. Pharmacol. 2009, 75, 1374–1379. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Cui, R.; Xu, X. miR-200c inhibits melanoma progression and drug resistance through down-regulation of BMI-1. Am. J. Pathol. 2012, 181, 1823–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryndyak, V.P.; Beland, F.A.; Pogribny, I.P. E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int. J. Cancer 2010, 126, 2575–2583. [Google Scholar] [CrossRef]
- Medarova, Z.; Pantazopoulos, P.; Yoo, B. creening of potential miRNA therapeutics for the prevention of multi-drug resistance in cancer cells. Sci. Rep. 2020, 10, 1970. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.J.; Huang, X. Ion Channels in Cancer: Orchestrators of Electrical Signaling and Cellular Crosstalk. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Capatina, A.L.; Lagos, D.; Brackenbury, W.J. Targeting Ion Channels for Cancer Treatment: Current Progress and Future Challenges. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Rodríguez-Rasgado, J.A.; Acuña-Macías, I.; Camacho, J. Eag1 channels as potential cancer biomarkers. Sensors 2012, 12, 5986–5995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlaz Us, S.; Sogut, F.; Yildirim, M.; Yetkin, D.; Yalin, S.; Yilmaz, S.N.; Comelekoglu, U. Effect of Imipramine on radiosensitivity of Prostate Cancer: An In Vitro Study. Cancer Investig. 2019, 37, 489–500. [Google Scholar] [CrossRef]
- Teschemacher, A.G.; Seward, E.P.; Hancox, J.C.; Witchel, H.J. Inhibition of the current of heterologously expressed HERG potassium channels by imipramine and amitriptyline. Br. J. Pharmacol. 1999, 128, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ferreiro, R.E.; Kerschensteiner, D.; Major, F.; Monje, F.; Stühmer, W.; Pardo, L.A. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J. Gen. Physiol. 2004, 124, 301–317. [Google Scholar] [CrossRef] [Green Version]
- Rajamanickam, S.; Panneerdoss, S.; Gorthi, A.; Timilsina, S.; Onyeagucha, B.; Kovalskyy, D.; Ivanov, D.; Hanes, M.A.; Vadlamudi, R.K.; Chen, Y.; et al. Inhibition of FoxM1-Mediated DNA Repair by Imipramine Blue Suppresses Breast Cancer Growth and Metastasis. Clin. Cancer Res. 2016, 22, 3524–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, E.; Hammadi, M.; Kischel, P.; Delcroix, V.; Demaurex, N.; Castelbou, C.; Vacher, A.M.; Devin, A.; Ducret, T.; Nunes, P.; et al. The antidepressant fluoxetine induces necrosis by energy depletion and mitochondrial calcium overload. Oncotarget 2017, 8, 3181–3196. [Google Scholar] [CrossRef]
- Stepulak, A.; Rzeski, W.; Sifringer, M.; Brocke, K.; Gratopp, A.; Kupisz, K.; Turski, L.; Ikonomidou, C. Fluoxetine inhibits the extracellular signal regulated kinase pathway and suppresses growth of cancer cells. Cancer Biol. Ther. 2008, 7, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Grygier, B.; Arteta, B.; Kubera, M.; Basta-Kaim, A.; Budziszewska, B.; Leśkiewicz, M.; Curzytek, K.; Duda, W.; Lasoń, W.; Maes, M. Inhibitory effect of antidepressants on B16F10 melanoma tumor growth. Pharmacol. Rep. 2013, 65, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Po, W.W.; Thein, W.; Khin, P.P.; Khing, T.M.; Han, K.; Park, C.H.; Sohn, U.D. Fluoxetine Simultaneously Induces Both Apoptosis and Autophagy in Human Gastric Adenocarcinoma Cells. Biomol. Ther. 2020, 28, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Xu, L.; Zhang, X.; Peng, W.; Tang, Q.; Feng, C. The proliferation effects of fluoxetine and amitriptyline on human breast cancer cells and the underlying molecular mechanisms. Environ. Toxicol. Pharmacol. 2021, 83, 103586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Z.; Gong, Q.; Makielski, J.C.; January, C.T. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ. Res. 1999, 84, 989–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.J.; Ma, J.H.; Zhang, P.H.; Wang, X.P.; Zou, A.R.; Tu, D.N. Verapamil blocks HERG channel by the helix residue Y652 and F656 in the S6 transmembrane domain. Acta Pharmacol. Sin. 2007, 28, 959–967. [Google Scholar] [CrossRef]
- Brocchieri, A.; Saporiti, A.; Moroni, M.; Porta, C.; Tua, A.; Grignani, G. Verapamil inhibits to different extents agonist−induced Ca2+ transients in human tumor cells and in vitro tumor cell growth. Invasion Metastasis. 1996, 16, 56–64. [Google Scholar]
- Taylor, J.M.; Simpson, R.U. Inhibition of cancer cell growth by calcium channel antagonists in the athymic mouse. Cancer Res. 1992, 52, 2413–2418. [Google Scholar] [PubMed]
- Li, P.; Zhong, D.; Gong, P.Y. Synergistic effect of paclitaxel and verapamil to overcome multi-drug resistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 516, 183–188. [Google Scholar] [CrossRef]
- Holdhoff, M.; Ye, X.; Supko, J.G.; Nabors, L.B.; Desai, A.S.; Walbert, T.; Lesser, G.J.; Read, W.L.; Lieberman, F.S.; Lodge, M.A.; et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro Oncol. 2017, 19, 845–852. [Google Scholar] [CrossRef] [Green Version]
- García-Quiroz, J.; García-Becerra, R.; Barrera, D.; Santos, N.; Avila, E.; Ordaz-Rosado, D.; Rivas-Suárez, M.; Halhali, A.; Rodríguez, P.; Gamboa-Domínguez, A.; et al. Astemizole synergizes calcitriol antiproliferative activity by inhibiting CYP24A1 and upregulating VDR: A novel approach for breast cancer therapy. PLoS ONE 2012, 7, e45063. [Google Scholar] [CrossRef]
- García-Quiroz, J.; García-Becerra, R.; Santos-Martínez, N.; Barrera, D.; Ordaz-Rosado, D.; Avila, E.; Halhali, A.; Villanueva, O.; Ibarra-Sánchez, M.J.; Esparza-López, J.; et al. In vivo dual targeting of the oncogenic Ether-à-go-go-1 potassium channel by calcitriol and astemizole results in enhanced antineoplastic effects in breast tumors. BMC Cancer 2014, 14, 745. [Google Scholar] [CrossRef] [Green Version]
- García-Quiroz, J.; García-Becerra, R.; Santos-Cuevas, C.; Ramírez-Nava, G.J.; Morales-Guadarrama, G.; Cárdenas-Ochoa, N.; Segovia-Mendoza, M.; Prado-Garcia, H.; Ordaz-Rosado, D.; Avila, E.; et al. Synergistic Antitumorigenic Activity of Calcitriol with Curcumin or Resveratrol is Mediated by Angiogenesis Inhibition in Triple Negative Breast Cancer Xenografts. Cancers 2019, 11, 1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo-Torres, E.; Rodríguez, G.; Meneses-Morales, I.; Zarain-Herzberg, A. ATP2A3 gene as an important player for resveratrol anticancer activity in breast cancer cells. Mol. Carcinog. 2017, 56, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Torres, E.; Hernández-Oliveras, A.; Meneses-Morales, I.; Rodríguez, G.; Fuentes-García, G.; Zarain-Herzberg, Á. Resveratrol up-regulates ATP2A3 gene expression in breast cancer cell lines through epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2019, 113, 37–47. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, M.; Gao, J.; Li, J.; Fan, L.; Xiang, G.; Wang, X.; Wang, X.; Wu, X.; Sun, Y.; et al. Small molecule RL71 targets SERCA2 at a novel site in the treatment of human colorectal cancer. Oncotarget 2015, 6, 37613–37625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase−2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, V.C.; Lung, S.S.; Pu, J.K.; Hung, K.N.; Leung, G.K. Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Res. 2010, 30, 4515–4524. [Google Scholar] [PubMed]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Tamborini, M.; Locatelli, E.; Rasile, M.; Monaco, I.; Rodighiero, S.; Corradini, I.; Franchini, M.C.; Passoni, L.; Matteoli, M. Combined Approach Employing Chlorotoxin-Nanovectors and Low Dose Radiation to Reach Infiltrating Tumor Niches in Glioblastoma. ACS Nano. 2016, 10, 2509–2520. [Google Scholar] [CrossRef]
- Dastpeyman, M.; Giacomin, P.; Wilson, D.; Nolan, M.J.; Bansal, P.S.; Daly, N.L. A C-Terminal Fragment of Chlorotoxin Retains Bioactivity and Inhibits Cell Migration. Front. Pharmacol. 2019, 10, 250. [Google Scholar] [CrossRef]
- Lefranc, F.; Le Rhun, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.O.; Ningaraj, N.S. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9, 258. [Google Scholar] [CrossRef] [Green Version]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2007, 26, 2525–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, A.; Reinhardt, J.; Schneider, S.W.; Gassner, B.; Schuricht, B. K (+) channel-dependent migration of fibroblasts and human melanoma cells. Cell. Physiol. Biochem. 1999, 9, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Pedarzani, P.; D’hoedt, D.; Doorty, K.B.; Wadsworth, J.D.; Joseph, J.S.; Jeyaseelan, K.; Kini, R.M.; Gadre, S.V.; Sapatnekar, S.M.; Stocker, M.; et al. a venom peptide from the Indian red scorpion (Mesobuthus tamulus) that targets small conductance Ca2+-activated K+ channels and afterhyperpolarization currents in central neurons. J. Biol. Chem. 2002, 277, 46101–46109. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Cordero, B.; Toledano, Y.; Cano-Sánchez, P.; Hernández-López, R.; Flores-Solis, D.; Saucedo-Yáñez, A.L.; Chávez-Uribe, I.; Brieba, L.G.; del Río-Portilla, F. Cytotoxicity of recombinant tamapin and related toxin-like peptides on model cell lines. Chem. Res. Toxicol. 2014, 27, 960–967. [Google Scholar] [CrossRef]
- Mayorga-Flores, M.; Chantôme, A.; Melchor-Meneses, C.M.; Domingo, I.; Titaux-Delgado, G.A.; Galindo-Murillo, R.; Vandier, C.; Del Río-Portilla, F. Novel Blocker of Onco SK3 Channels Derived from Scorpion Toxin Tamapin and Active against Migration of Cancer Cells. ACS Med. Chem. Lett. 2020, 11, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Duranti, C.; Arcangeli, A. Ion Channel Targeting with Antibodies and Antibody Fragments for Cancer Diagnosis. Antibodies 2019, 8, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.M.; Gidley Baird, A.; Glazer, S.; Barden, J.A.; Glazer, A.; Teh, L.C.; King, J. A phase I clinical trial demonstrates that nfP2 × 7 -targeted antibodies provide a novel, safe and tolerable topical therapy for basal cell carcinoma. Br. J. Dermatol. 2017, 177, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.M.; Oliphant, C.J.; Hassan, S.; Peille, A.L.; Bronsert, P.; Falzoni, S.; Di Virgilio, F.; McNulty, S.; Lara, R. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 2019, 38, 194–208. [Google Scholar] [CrossRef] [Green Version]
- Hirschler-Laszkiewicz, I.; Chen, S.J.; Bao, L.; Wang, J.; Zhang, X.Q.; Shanmughapriya, S.; Keefer, K.; Madesh, M.; Cheung, J.Y.; Miller, B.A. The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation. Am. J. Physiol. Cell Physiol. 2018, 315, C571–C586. [Google Scholar] [CrossRef]
- Chen, S.J.; Bao, L.; Keefer, K.; Shanmughapriya, S.; Chen, L.; Lee, J.; Wang, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Merali, S.; et al. Transient receptor potential ion channel TRPM2 promotes AML proliferation and survival through modulation of mitochondrial function, ROS, and autophagy. Cell Death Dis. 2020, 11, 247. [Google Scholar] [CrossRef] [Green Version]
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sánchez, S.; El Hiani, Y. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef] [Green Version]
- Almasi, S.; Long, C.Y.; Sterea, A.; Clements, D.R.; Gujar, S.; El Hiani, Y. TRPM2 Silencing Causes G2/M Arrest and Apoptosis in Lung Cancer Cells via Increasing Intracellular ROS and RNS Levels and Activating the JNK Pathway. Cell. Physiol. Biochem. 2019, 52, 742–757. [Google Scholar] [PubMed]
- Sales, T.T.; Resende, F.F.; Chaves, N.L.; Titze-De-Almeida, S.S.; Báo, S.N.; Brettas, M.L.; Titze-De-Almeida, R. Suppression of the Eag1 potassium channel sensitizes glioblastoma cells to injury caused by temozolomide. Oncol. Lett. 2016, 12, 2581–2589. [Google Scholar] [CrossRef]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Liao, H.; Liu, T.; Zeng, X.; Xiao, F.; Luo, L.; Guo, H.; Guo, L. MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (EAG1). Eur. J. Cancer 2013, 49, 710–724. [Google Scholar] [CrossRef]
- Shi, X.; He, W.; Guo, S.; Zhang, B.; Ren, S.; Liu, K.; Sun, T.; Cui, J. RNA-seq Analysis of the SCN1A-KO Model based on CRISPR/Cas9 Genome Editing Technology. Neuroscience 2019, 398, 1–11. [Google Scholar] [CrossRef]
- Chai, S.; Kan, S.; Sun, R.; Zhou, R.; Sun, Y.; Chen, W.; Yu, B. Fabricating polydopamine-coated MoSe2-wrapped hollow mesoporous silica nanoplatform for controlled drug release and chemo-photothermal therapy. Int. J. Nanomed. 2018, 13, 7607–7621. [Google Scholar] [CrossRef] [Green Version]
- Kulbacka, J.; Chodaczek, G.; Rossowska, J.; Szewczyk, A.; Saczko, J.; Bazylińska, U. Investigating the photodynamic efficacy of chlorin e6 by millisecond pulses in metastatic melanoma cells. Bioelectrochemistry 2020, 138, 107728. [Google Scholar] [CrossRef] [PubMed]
- Buckner, C.A.; Buckner, A.L.; Koren, S.A.; Persinger, M.A.; Lafrenie, R.M. Inhibition of cancer cell growth by exposure to a specific time-varying electromagnetic field involves T-type calcium channels. PLoS ONE 2015, 10, e0124136. [Google Scholar] [CrossRef] [PubMed]
- Wust, P.; Kortüm, B.; Strauss, U.; Nadobny, J.; Zschaeck, S.; Beck, M.; Stein, U.; Ghadjar, P. Non-thermal effects of radiofrequency electromagnetic fields. Sci. Rep. 2020, 10, 13488. [Google Scholar] [CrossRef]
- Heldin, C.H.; Lu, B.; Evans, R.; Gutkind, J.S. Signals and Receptors. Cold Spring Harb. Perspect. Biol. 2016, 8, a005900. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocek, E.; Moulas, A.N.; Studzinski, G.P. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit. Rev. Clin. Lab. Sci. 2014, 51, 125–137. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 625–1637. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor-tyrosine kinases. Cell 2010, 25, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Shi, Q.; West, T.M.; Xiang, Y.K. Cross−Talk Between Insulin Signaling and G Protein-Coupled Receptors. J. Cardiovasc. Pharmacol. 2017, 70, 74–86. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Receptor heterodimerization: A new level of cross−talk. J. Clin. Investig. 2006, 116, 1210–1212. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef] [Green Version]
- Aittaleb, M.; Boguth, C.A.; Tesmer, J.J. Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 2010, 77, 111–125. [Google Scholar] [CrossRef] [Green Version]
- Grusch, M.; Petz, M.; Metzner, T.; Oztürk, D.; Schneller, D.; Mikulits, W. The crosstalk of RAS with the TGF-β family during carcinoma progression and its implications for targeted cancer therapy. Curr. Cancer Drug. Targets 2010, 10, 849–857. [Google Scholar] [CrossRef]
- Mendelson, K.; Swendeman, S.; Saftig, P.; Blobel, C.P. Stimulation of platelet-derived growth factor receptor beta (PDGFRbeta) activates ADAM17 and promotes metalloproteinase-dependent cross−talk between the PDGFRbeta and epidermal growth factor receptor (EGFR) signaling pathways. J. Biol. Chem. 2010, 285, 25024–25032. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.J.; Miller, L.L. Changes in the plasma membrane in metabolic disease: Impact of the membrane environment on G protein-coupled receptor structure and function. Br. J. Pharmacol. 2018, 175, 4009–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbro, D.; Cowan-Jacob, S.W.; Möbitz, H.; Martiny-Baron, G. Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol. Biol. 2012, 795, 1–34. [Google Scholar] [PubMed]
- Xie, Y.H.; Chen, Y.H.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [PubMed]
- Du, Z.; Lovely, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Drescher, D.; Moehler, M.; Gockel, I.; Frerichs, K.; Müller, A.; Dünschede, F.; Borschitz, T.; Biesterfeld, S.; Holtmann, M.; Wehler, T.; et al. Coexpression of receptor-tyrosine-kinases in gastric adenocarcinoma--a rationale for a molecular targeting strategy? World J. Gastroenterol. 2007, 26, 3605–3609. [Google Scholar] [CrossRef] [Green Version]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer therapy International. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasota, J.; Miettinen, M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin. Diagn. Pathol. 2006, 23, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toffalini, F.; Kallin, A.; Vandenberghe, P.; Pierre, P.; Michaux, L.; Cools, J.; Demoulin, J.B. The fusion proteins TEL-PDGFRbeta and FIP1L1-PDGFRalpha escape ubiquitination and degradation. Haematologica 2009, 94, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef] [Green Version]
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell 2002, 1, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Gibson, T.B.; Ranganathan, A.; Grothey, A. Randomized phase III trial results of panitumumab, a fully human anti-epidermal growth factor receptor monoclonal antibody, in metastatic colorectal cancer. Clin. Colorectal Cancer 2006, 6, 29–31. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E.J.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. APHINITY Steering Committee and Investigators. Adjuvant Pertuzumab and Trastuzumab in early HER2-positive breast cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Prechtel, D.; Resau, J.H.; Gauger, K.; Welk, A.; Lindemann, K.; Salanti, G.; Richter, T.; Knudsen, B.; Vande Woude, G.F.; et al. C-met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of her2/neu. Int. J. Cancer 2005, 113, 678–682. [Google Scholar] [CrossRef]
- Vigh, L.; Escribá, P.V.; Sonnleitner, A.; Sonnleitner, M.; Piotto, S.; Maresca, B.; Horváth, I.; Harwood, J.L. The significance of lipid composition for membrane activity: New concepts and ways of assessing function. Prog. Lipid Res. 2005, 44, 303–344. [Google Scholar] [CrossRef]
- Frewein, M.; Kollmitzer, B.; Heftberger, P.; Pabst, G. Lateral pressure-mediated protein partitioning into liquid-ordered/liquid-disordered domains. Soft Matter. 2016, 12, 3189–3195. [Google Scholar] [CrossRef] [Green Version]
- Ghysels, A.; Krämer, A.; Venable, R.M.; Teague, W.E., Jr.; Lyman, E.; Gawrisch, K.; Pastor, R.W. Permeability of membranes in the liquid ordered and liquid disordered phases. Nat. Commun. 2019, 10, 5616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in cancer: The relevance of drug-membrane interaction studies. Biochim. Biophys. Acta 2016, 1858, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.; Luberto, C.; Canals, D. Inhibitors of the sphingomyelin cycle: Sphingomyelin synthases and sphingomyelinases. Chem. Phys. Lipids 2016, 197, 45–59. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Horta, M.; Lima, J.; Nunes, C.; Reis, S. A biophysical approach to daunorubicin interaction with model membranes: Relevance for the drug’s biological activity. J. R. Soc. Interface 2017, 14, 20170408. [Google Scholar] [CrossRef] [PubMed]
- Barceló-Coblijn, G.; Martin, M.L.; de Almeida, R.F.; Noguera-Salvà, M.A.; Marcil-la-Etxenike, A.; Guardiola-Serrano, F.; Lüth, A.; Kleuser, B.; Halver, J.E.; Escribá, P.V. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. USA 2011, 6, 19569–19574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [Green Version]
- Hao, M.; Mukherjee, S.; Sun, Y.; Maxfield, F.R. Effects of cholesterol depletion and increased lipid unsaturation on the properties of endocytic membranes. J. Biol. Chem. 2004, 279, 14171–14178. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, R.A.; Zerouga, M.; Wu, M.; Castillo, A.; Harvey, K.; Zaloga, G.P.; Stillwell, W. Anticancer properties of propofol-docosahexaenoate and propofol-eicosapentaenoate on breast cancer cells. Breast Cancer Res. 2005, 7, R645. [Google Scholar] [CrossRef]
- Lajoie, P.; Nabi, I.R. Lipid rafts, caveolae, and their endocytosis. Int. Rev. Cell Mol. Biol. 2010, 282, 135–163. [Google Scholar]
- Huang, Q.; Shen, H.-M.; Shui, G.; Wenk, M.R.; Ong, C.-N. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid raft-associated integrin signaling pathway. Cancer Res. 2006, 66, 5807–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.-I.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Luit, A.H.; Budde, M.; Ruurs, P.; Verheij, M.; van Blitterswijk, W.J. Alkyl-lysophospholipid accumulates in lipid rafts and induces apoptosis via raft-dependent endocytosis and inhibition of phosphatidylcholine synthesis. J. Biol. Chem. 2002, 18, 39541–39547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczewska, A.; Dziedzic, B.; Stulczewski, D.; Zgórzyńska, E. Cell membranes. Molecular lipid therapy. Postepy Hig. Med. Dosw 2017, 71, 1239–1250. [Google Scholar] [CrossRef]
- Delmas, D.; Aires, V.; Colin, D.J.; Limagne, E.; Scagliarini, A.; Cotte, A.K.; Ghiringhelli, F. Importance of lipid microdomains, rafts, in absorption, delivery, and biological effects of resveratrol. Ann. N. Y. Acad. Sci. 2013, 1290, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Böckmann, R.A.; De Groot, B.L.; Kakorin, S.; Neumann, E.; Grubmüller, H. Kinetics, statistics, and energetics of lipid membrane electroporation studied by molecular dynamics simulations. Biophys. J. 2008, 95, 1837–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotnik, T.; Rems, L.; Tarek, M.; Miklavcic, D. Membrane Electroporation and Electropermeabilization: Mechanisms and Models. Annu. Rev. Biophys. 2019, 48, 63–91. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, T.R.; Esser, A.T.; Vasilkoski, Z.; Smith, K.C.; Weaver, J.C. Microdosimetry for conventional and supra-electroporation in cells with organelles. Biochem. Biophys. Res. Commun. 2006, 341, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Saulis, G.; Saulė, R. Size of the pores created by an electric pulse: Microsecond vs. millisecond pulses. Biochim. Biophys. Acta 2012, 1818, 3032–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieleman, D.P. The molecular basis of electroporation. BMC Biochem. 2004, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benov, L.C.; Antonov, P.A.; Ribarov, S.R. Oxidative damage of the membrane lipids after electroporation. Gen. Physiol. Biophys. 1994, 13, 85–97. [Google Scholar] [PubMed]
- Gabriel, B.; Teissié, J. Generation of reactive-oxygen species induced by electropermeabilization of Chinese hamster ovary cells and their consequence on cell viability. Eur. J. Biochem. 1994, 223, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bladergroen, M.R.; Rosato, N.; Agro, A.F. Role of lipid peroxidation in electroporation-induced cell permeability. Biochem. Biophys. Res. Commun. 1995, 209, 417–425. [Google Scholar] [CrossRef]
- Tavazzi, B.; Di, P.D.; Amorini, A.M.; Fazzina, G.; Tuttobene, M.; Giardina, B.; Lazzarino, G. Energy metabolism and lipid peroxidation of human erythrocytes as a function of increased oxidative stress. Eur. J. Biochem. 2000, 267, 684–689. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Min, B.; Ahn, D. Mechanism of lipid peroxidation in meat and meat products-a review. Food Sci. Biotechnol. 2005, 14, 152–163. [Google Scholar]
- Fortier, C.A.; Guan, B.; Cole, R.B.; Tarr, M.A. Covalently bound fluorescent probes as reporters for hydroxyl radical penetration into liposomal membranes. Free Radic. Biol. Med. 2009, 46, 1376–1385. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.S.; Lander, A.; Darley-Usmar, V. Mitochondrial proteomics in free radical research. Free Rad. Biol. Med. 2005, 38, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Vernier, P.T.; Sun, Y.H.; Gundersen, M.A. Nanoelectropulse-driven membrane perturbation and small molecule permeabilization. BMC Cell Biol. 2006, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakhomov, A.G.; Kolb, J.F.; White, J.A.; Joshi, R.P.; Xiao, S.; Schoenbach, K.H. Long-lasting plasma membrane permeabilization in mammalian cells by nanosecond pulsed electric field (nsPEF). Bioelectromagnetics 2007, 28, 655–663. [Google Scholar] [CrossRef]
- Hojman, P.; Gissel, H.; Andre, F.M.; Cournil-Henrionnet, C.; Eriksen, J.; Gehl, J.; Mir, L.M. Physiological effects of high and low voltage pulse combinations for gene electrotransfer in muscle. Hum. Gene Ther. 2008, 19, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Vernier, P.T.; Levine, Z.A.; Wu, Y.H.; Joubert, V.; Ziegler, M.J.; Mir, L.M.; Tieleman, D.P. Electroporating fields target oxidatively damaged areas in the cell membrane. PLoS ONE 2009, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bonnafous, P.; Vernhes, M.C.; Teissie, J.; Gabriel, B. The generation of reactive-oxygen species associated with long-lasting pulse-induced electropermeabilization of mammalian cells is based on a non-destructive alteration of the plasma membrane. Biochim. Biophys. Acta-Biomembr. 1999, 1461, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Sakharov, D.V.; Elstak, E.D.R.; Chernyak, B.; Wirtz, K.W.A. Prolonged lipid oxidation after photodynamic treatment. Study with oxidation-sensitive probe c11-bodipy581/591. FEBS Lett. 2005, 579, 1255–1260. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Berry, C.K.; Storer, P.A.; Raphael, R.M. Peroxidation of polyunsaturated phosphatidyl-choline lipids during electroformation. Biomaterials 2007, 28, 1298–1306. [Google Scholar] [CrossRef]
- Shevchuk, I.N.; Chekulayev, V.A.; Checulayeva, L.V. The role of lipid peroxidation and protein degradation in the photodestruction of Ehrlich ascites carcinoma cells sensitized by hemathoporphirin derivative. Exp. Oncol. 2002, 24, 216–224. [Google Scholar]
- Biedinger, U.; Youngman, R.J.; Schnabl, H. Differential effects of electrofusion and electropermeabilization parameters on the membrane integrity of plant protoplasts. Planta 1990, 180, 598–602. [Google Scholar] [CrossRef]
- Yeo, S.K.; Liong, M.T. Effect of electroporation on viability and bioconversion of isoflavones in mannitol-soymilk fermented by lactobacilli and bifidobacteria. J. Sci. Food Agric. 2013, 93, 396–409. [Google Scholar] [CrossRef]
- Yun, O.; Zeng, X.A.; Brennan, C.S.; Han, Z. Effect of pulsed electric field on membrane lipids and oxidative injury of Salmonella typhimurium. Int. J. Mol. Sci. 2016, 17, 1374. [Google Scholar] [CrossRef] [Green Version]
- Maccarrone, M.; Rosato, N.; Agro, A.F. Electroporation enhances cell membrane peroxidation and luminescence. Biochem. Biophys. Res. Commun. 1995, 206, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Boonnoy, P.; Jarerattanachat, V.; Karttunen, M.; Wong-Ekkabut, J. Bilayer deformation, pores, and micellation induced by oxidized lipids. J. Phys. Chem. Lett. 2015, 6, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Breton, M.; Delemotte, L.; Silve, A.; Mir, L.M.; Tarek, M. Transport of siRNA through lipid membranes driven by nanosecond electric pulses: An experimental and computational study. J. Am. Chem. Soc. 2012, 134, 13938–13941. [Google Scholar] [CrossRef]
- Nidernhofer, L.J.; Daniels, J.S.; Rouzer, C.A.; Greene, R.E.; Marnett, L.J. Malonodialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J. Biol. Chem. 2003, 278, 31426–31433. [Google Scholar] [CrossRef] [Green Version]
- Pakhomova, O.N.; Khorokhorina, V.A.; Bowman, A.M.; Rodaitė-Riševičienė, R.; Saulis, G.; Xiao, S.; Pakhomov, A.G. Oxidative effects of nanosecond pulsed electric field exposure in cells and cell-free media. Arch. Biochem. Biophys 2012, 527, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsong, T.Y. On electroporation of cell membranes and some related phenomena. Bioelectrochem. Bioenerg. 1990, 24, 271–295. [Google Scholar] [CrossRef]
- Tsong, T.Y. Electroporation of cell membranes. Biophys. J. 1991, 60, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.C.; Bardet, S.M.; Carr, L.; Romanenko, S.; Arnaud-Cormos, D.; Leveque, P.; O’Connor, R.P. Nanosecond pulsed electric fields depolarize transmembrane potential via voltage-gated K+, Ca2+ and TRPM8 channels in U87 glioblastoma cells. Biochim. Biophys. Acta 2017, 1859, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhongsheng, Z.; Lee, R.C. Supramembrane potential-induced electroconformational changes in sodium channel proteins: A potential mechanism involved in electric injury. Burns 2006, 32, 52–59. [Google Scholar] [CrossRef]
- Nesin, V.; Bowman, A.M.; Xiao, S.; Pakhomov, A.G. Cell permeabilization and inhibition of voltage gated Ca2+ and Na+ channel currents by nanosecond pulsed electric field. Bioelectromagnetics 2012, 33, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Nesin, V.; Pakhomov, A.G. Inhibition of voltage-gated Na+ current by nanosecond electric field (nsPEF) is not mediated by Na+ influx or Ca2+ signaling. Bioelectromagnetics 2012, 33, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Pakhomov, A.G.; Semenov, I.; Casciola, M.; Xiao, S. Neuronal excitation and permeabilization by 200-ns pulsed electric field: An optical membrane potential study with FluoVolt dye. Biochim. Biophys. Acta 2017, 1859, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Craviso, G.L.; Vernier, P.T.; Chatterjee, I.; Leblanc, N. Nanosecond electric pulses differentially affect inward and outward currents in patch clamped adrenal chromaffin cells. PLoS ONE 2017, 12, e181002. [Google Scholar] [CrossRef]
- Teissié, J.; Tsong, T.Y. Evidence of voltage-induced channel opening in Na/K ATPase of human erythrocyte membrane. J. Membr. Biol. 1980, 55, 133–1340. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 1, a018267. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, G.L.; Roth, C.; Tolstykh, G.; Kuipers, M.; Ibey, B.L. Disruption of the actin cortex contributes to susceptibility of mammalian cells to nanosecond pulsed electric fields. Bioelectromagnetics 2014, 35, 262–272. [Google Scholar] [CrossRef]
- Stacey, M.; Fox, P.; Buescher, S.; Kolb, J. Nanosecond pulsed electric field induced cytoskeleton, nuclear membrane and telomere damage adversely impact cell survival. Bioelectrochemistry 2011, 82, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Pehlivanova, V.N.; Tsoneva, I.H.; Tzoneva, R.D. Multiple effects of electroporation on the adhesive behaviour of breast cancer cells and fibroblasts. Cancer Cell Int. 2012, 12, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, R.M. Reactive oxygen species at phospholipid bilayers: Distribution, mobility and permeation. Biochim. Biophys. Acta Biomembr. 2014, 1838, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napotnik, T.B.; Reberšek, M.; Vernier, P.T.; Mali, B.; Miklavčič, D. Effects of high voltage nanosecond electric pulses on eucaryotic cells (in vitro): A systematic review. Bioelectrochemistry 2016, 110, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuer, A.; Wende, K.; Babica, P.; Kolb, J.B. Elasticity and tumorigenic characteristics of cells in a monolayer after nanosecond pulsed electric field exposure. Eur. Biophys. J. 2017, 46, 567–580. [Google Scholar] [CrossRef]
- Szewczyk, A.; Gehl, J.; Daczewska, M.; Saczko, J.; Frandsen, S.K.; Kulbacka, J. Calcium electroporation for treatment of sarcoma in preclinical studies. Oncotarget 2018, 14, 11604–11618. [Google Scholar] [CrossRef] [Green Version]
- Graybill, P.M.; Davalos, R.V. Cytoskeletal disruption after electroporation and its significance to pulsed electric field therapies. Cancers 2020, 12, 1132. [Google Scholar] [CrossRef] [PubMed]
- Kollman, J.M.; Merdes, A.; Mourey, L.; Agard, D.A. Microtubule nucleation by γ-tubulin complexes. Nat. Rev. Mol. Cell Biol. 2011, 12, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Harkin, F.G.; Hay, E.D. Effects of electroporation on the tubulin cytoskeleton and directed migration of corneal fibroblasts cultured within collagen matrices. Cell Motil. Cytoskelet. 1996, 35, 345–357. [Google Scholar] [CrossRef]
- Meulenberg, C.J.W.; Todorovic, V.; Cemazar, M. Differential Cellular Effects of Electroporation and Electrochemotherapy in Monolayers of Human Microvascular Endothelial Cells. PLoS ONE 2012, 12, e52713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanthou, C.; Kranjc, S.; Sersa, G.; Tozer, G.; Zupanic, A.; Cemazar, M. The endothelial cytoskeleton as a target of electroporation-based therapies. Mol. Cancer Ther. 2006, 12, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Carr, L.; Bardet, S.M.; Burke, R.C.; Arnaud-Cormos, D.; Leveque, P.; O’Connor, R.P. Calcium-independent disruption of microtubule dynamics by nanosecond pulsed electric fields in U87 human glioblastoma cells. Sci. Rep. 2017, 7, 41267. [Google Scholar] [CrossRef]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate filaments at the junction of mechanotransduction, migration, and development. Front. Cell Dev. Biol. 2017, 5, 81. [Google Scholar] [CrossRef]
- Thompson, G.L.; Roth, C.C.; Kuipers, M.A.; Tolstykh, G.P.; Beier, H.T.; Ibey, B.L. Permeabilization of the nuclear envelope following nanosecond pulsed electric field exposure. Biochem. Biophys. Res. Commun. 2016, 470, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmush, M.L.; Golberg, A.; Serša, G.; Kotnik, T.; Miklavčič, D. Electroporation-based technologies for medicine: Principles, applications, and challenges. Annu. Rev. Biomed. Eng. 2014, 16, 295–320. [Google Scholar] [CrossRef] [Green Version]
- Giardino, R.; Fini, M.; Bonazzi, V.; Cadossi, R.; Nicolini, A.; Carpi, A. Electrochemotherapy a novel approach to the treatment of metastatic nodules on the skin and subcutaneous tissues. Biomed. Pharmacother. 2006, 60, 458–462. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, Y.; Huang, J.; Wang, Y.; Rubinsky, B. Study on Nonthermal Irreversible Electroporation of the Thyroid. Technol. Cancer Res. Treat. 2019, 18, 1533033819876307. [Google Scholar] [CrossRef] [PubMed]
- Cemazar, M.; Jarm, T.; Sersa, G. Cancer electrogene therapy with interleukin-12. Curr. Gene Ther. 2010, 10, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Rosati, M.; Valentin, A.; Jalah, R.; Patel, V.; von Gegerfelt, A.; Bergamaschi, C.; Alicea, C.; Weiss, D.; Treece, J.; Pal, R.; et al. Increased immune responses in rhesus macaques by DNA vaccination combined with electroporation. Vaccine 2008, 26, 5223–5229. [Google Scholar] [CrossRef]
- Ward, M.; Wu, J. Ultrasound-induced Cell Lysis and Sonoporation Enhanced by Contrast Agents. J. Acoust. Soc. Am. 1999, 105, 2951–2957. [Google Scholar] [CrossRef] [PubMed]
- Escoffre, J.M.; Novell, A.; Piron, J.; Zeghimi, A.; Doinikov, A.; Bouakaz, A. Microbubble Attenuation and Destruction: Are They Involved in Sonoporation Efficiency? IEEE Trans. Ultrason. Ferroelectr. Freq. Control. 2013, 60, 46–52. [Google Scholar] [CrossRef]
- van Wamel, A.; Kooiman, K.; Harteveld, M.; Emmer, M.; ten Cate, F.J.; Versluis, M.; de Jong, N. Vibrating Microbubbles Poking Individual Cells: Drug Transfer into Cells via Sonoporation. J. Control. Release 2006, 112, 149–155. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Q.; Guo, X.; Tu, J.; Zhang, D. Mechanisms Underlying Sonoporation: Interaction between Microbubbles and Cells. Ultrason. Sonochem. 2020, 67, 105096. [Google Scholar] [CrossRef]
- Lentacker, I.; De Cock, I.; Deckers, R.; De Smedt, S.C.; Moonen, C.T.W. Understanding Ultrasound Induced Sonoporation: Definitions and Underlying Mechanisms. Adv. Drug Deliv. Rev. 2014, 72, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooiman, K.; Vos, H.J.; Versluis, M.; De Jong, N. Acoustic Behavior of Microbubbles and Implications for Drug Delivery. Adv. Drug Deliv. Rev. 2014, 72, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Bouakaz, A.; Versluis, M.; De Jong, N. High-Speed Optical Observations of Contrast Agent Destruction. Ultrasound Med. Biol. 2005, 31, 391–399. [Google Scholar] [CrossRef]
- Wu, J.; Nyborg, W.L. Ultrasound, Cavitation Bubbles and Their Interaction with Cells. Adv. Drug Deliv. Rev. 2008, 60, 1103–1116. [Google Scholar] [CrossRef]
- Helfield, B.; Chen, X.; Watkins, S.C.; Villanueva, F.S. Biophysical Insight into Mechanisms of Sonoporation. Proc. Natl. Acad. Sci. USA 2016, 113, 9983–9988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, D.; Wong, J.; Griffin, B.; Ellis, S.G.; Porter, T.; Sen, S.; Thomas, J.D. Ten-Fold Augmentation of Endothelial Uptake of Vascular Endothelial Growth Factor with Ultrasound after Systemic Administration. J. Am. Coll. Cardiol. 2000, 35, 1678–1686. [Google Scholar] [CrossRef] [Green Version]
- Zderic, V.; Clark, J.I.; Martin, R.W.; Vaezy, S. Ultrasound-Enhanced Transcorneal Drug Delivery. Cornea 2004, 23, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Huber, P.E.; Pfisterer, P. In Vitro and in Vivo Transfection of Plasmid DNA in the Dunning Prostate Tumor R3327-AT1 Is Enhanced by Focused Ultrasound. Gene Ther. 2000, 17, 1516–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.R. Ultrasonic Exposure Thresholds for Changes in Cells and Tissues. J. Acoust. Soc. Am. 1971, 90, 1971. [Google Scholar] [CrossRef]
- Mitome, H. The Mechanism of Generation of Acoustic Streaming. Electron. Commun. Jpn. 1998, 81, 1–8. [Google Scholar] [CrossRef]
- Zhang, C.B.; Liu, Z.; Guo, X.S.; Zhang, D. Correlation between Microbubble-Induced Acoustic Cavitation and Hemolysis in Vitro. Chin. Phys. B 2011, 20, 024301. [Google Scholar] [CrossRef]
- Tran, B.C.; Seo, J.; Hall, T.L.; Fowlkes, J.B.; Cain, C.A. Microbubble-Enhanced Cavitation for Noninvasive Ultrasound Surgery. IEEE Trans. Ultrason. Ferroelectr. Freq. Control. 2003, 50, 1296–1304. [Google Scholar] [CrossRef]
- Kimmel, E. Cavitation Bioeffects. Crit. Rev. Biomed. Eng. 2006, 34, 105–161. [Google Scholar] [CrossRef] [PubMed]
- Udroiu, I. Ultrasonic Drug Delivery in Oncology. J. BUON 2015, 20, 381–390. [Google Scholar]
- Jelenc, J.; Jelenc, J.; Miklavčič, D.; Lebar, A.M. Low-Frequency Sonoporation in Vitro: Experimental System Evaluation. J. Mech. Eng. 2012, 58, 319–326. [Google Scholar] [CrossRef]
- Liu, Z.; You, H.; Zhang, P.; Lin, R.; Chang, Y. Thermal Simulation and Sonoporation Experiment Based on a Focused Ultrasonic System. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Cairo, Egypt, 7–9 April 2020; Volume 739, p. 012037. [Google Scholar]
- Jiang, X.; Savchenko, O.; Li, Y.; Qi, S.; Yang, T.; Zhang, W.; Chen, J. A Review of Low-Intensity Pulsed Ultrasound for Therapeutic Applications. IEEE Trans. Biomed. Eng. 2019, 66, 2704–2718. [Google Scholar] [CrossRef]
- Burgess, M.T.; Porter, T.M. Control of Acoustic Cavitation for Efficient Sonoporation with Phase-Shift Nanoemulsions. Ultrasound Med. Biol. 2019, 45, 846–858. [Google Scholar] [CrossRef]
- Sundaram, J.; Mellein, B.R.; Mitragotri, S. An Experimental and Theoretical Analysis of Ultrasound-Induced Permeabilization of Cell Membranes. Biophys. J. 2003, 84, 3087–3101. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhang, Y.; Cai, C.; Tu, J.; Guo, X.; Zhang, D. Sonoporation-Induced Cell Membrane Permeabilization and Cytoskeleton Disassembly at Varied Acoustic and Microbubble-Cell Parameters. Sci. Rep. 2018, 8, 3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bai, W.K.; Shen, E.; Hu, B. Sonoporation by Low-Frequency and Low-Power Ultrasound Enhances Chemotherapeutic Efficacy in Prostate Cancer Cells in Vitro. Oncol. Lett. 2013, 6, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.L.; Pislaru, S.V.; Greenleaf, J.F. Sonoporation: Mechanical DNA Delivery by Ultrasonic Cavitation. Somat. Cell Mol. Genet. 2002, 27, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.D.; Tang, J.; Halliwell, M. Sonoporation, Drug Delivery, and Gene Therapy. Proc. Inst. Mech. Eng. Part H 2010, 224, 343–361. [Google Scholar] [CrossRef]
- Belling, J.N.; Heidenreich, L.K.; Tian, Z.; Mendoza, A.M.; Chiou, T.T.; Gong, Y.; Chen, N.Y.; Young, T.D.; Wattanatorn, N.; Park, J.H. Acoustofluidic Sonoporation for Gene Delivery to Human Hematopoietic Stem and Progenitor Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 10976–10982. [Google Scholar] [CrossRef]
- Dimcevski, G.; Kotopoulis, S.; Bjånes, T.; Hoem, D.; Schjøt, J.; Gjertsen, B.T.; Biermann, M.; Molven, A.; Sorbye, H.; McCormack, E.; et al. Human Clinical Trial Using Ultrasound and Microbubbles to Enhance Gemcitabine Treatment of Inoperable Pancreatic Cancer. J. Control. Release 2016, 243, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.G.; Berry, J.L.; Lee, T.C.; Wang, A.T.; Honowitz, S.; Linn Murphree, A.; Varshney, N.; Hinton, D.R.; Fawzi, A.A. Sonoporation Enhances Chemotherapeutic Efficacy in Retinoblastoma Cells in Vitro. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3868–3873. [Google Scholar] [CrossRef]
- Hirabayashi, F.; Iwanaga, K.; Okinaga, T.; Takahashi, O.; Ariyoshi, W.; Suzuki, R.; Sugii, M.; Maruyama, K.; Tominaga, K.; Nishihara, T. Epidermal Growth Factor Receptor-Targeted Sonoporation with Microbubbles Enhances Therapeutic Efficacy in a Squamous Cell Carcinoma Model. PLoS ONE 2017, 12, e0185293. [Google Scholar] [CrossRef] [Green Version]
- Qu, N.; Shi, D.; Shang, M.; Duan, S.; Guo, L.; Ning, S.; Li, J. Breast Cancer Cell Line Phenotype Affects Sonoporation Efficiency under Optimal Ultrasound Microbubble Conditions. Med. Sci. Monit. 2018, 24, 9054–9062. [Google Scholar] [CrossRef]
- Shi, D.; Guo, L.; Duan, S.; Shang, M.; Meng, D.; Cheng, L.; Li, J. Influence of Tumor Cell Lines Derived from Different Tissue on Sonoporation Efficiency under Ultrasound Microbubble Treatment. Ultrason. Sonochem. 2017, 38, 598–603. [Google Scholar] [CrossRef]
- Theek, B.; Baues, M.; Ojha, T.; Möckel, D.; Veettil, S.K.; Steitz, J.; Van Bloois, L.; Storm, G.; Kiessling, F.; Lammers, T. Sonoporation Enhances Liposome Accumulation and Penetration in Tumors with Low EPR. J. Control. Release 2016, 231, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotopoulis, S.; Delalande, A.; Popa, M.; Mamaeva, V.; Dimcevski, G.; Gilja, O.H.; Postema, M.; Gjertsen, B.T.; McCormack, E. Sonoporation-Enhanced Chemotherapy Significantly Reduces Primary Tumour Burden in an Orthotopic Pancreatic Cancer Xenograft. Mol. Imaging Biol. 2014, 16, 53–62. [Google Scholar] [CrossRef]
- Zolochevska, O.; Xia, X.; Williams, B.J.; Ramsay, A.; Li, S.; Figueiredo, M.L. Sonoporation Delivery of Interleukin-27 Gene Therapy Efficiently Reduces Prostate Tumor Cell Growth in Vivo. Hum. Gene Ther. 2011, 22, 1537–1550. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Han, J.; Park, S.Y.; Kim, H.; Park, J.H.; Lee, H.J. Antitumor Efficacy of Focused Ultrasound-MFL Nanoparticles Combination Therapy in Mouse Breast Cancer Xenografts. Materials 2020, 13, 1099. [Google Scholar] [CrossRef] [Green Version]
- Elhelf, I.A.S.; Albahar, H.; Shah, U.; Oto, A.; Cressman, E.; Almekkawy, M. High Intensity Focused Ultrasound: The Fundamentals, Clinical Applications and Research Trends. Diagn. Interv. Imaging 2018, 99, 349–359. [Google Scholar] [CrossRef]
- Dubinsky, T.J.; Cuevas, C.; Dighe, M.K.; Kolokythas, O.; Joo, H.H. High-Intensity Focused Ultrasound: Current Potential and Oncologic Applications. Am. J. Roentgenol. 2008, 190, 191–199. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.B.; Chen, W.Z.; Wang, W.; Gui, Y.; Zhang, M.; Zheng, G.; Zhou, Y.; Xu, G.; Li, M.; et al. Extracorporeal High Intensity Focused Ultrasound Ablation in the Treatment of 1038 Patients with Solid Carcinomas in China: An Overview. Ultrason. Sonochem. 2004, 11, 149–154. [Google Scholar] [CrossRef]
- Kennedy, J.E.; Wu, F.; Ter Haar, G.R.; Gleeson, F.V.; Phillips, R.R.; Middleton, M.R.; Cranston, D. High-Intensity Focused Ultrasound for the Treatment of Liver Tumours. Ultrasonics 2004, 42, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-F. High Intensity Focused Ultrasound in Clinical Tumor Ablation. World J. Clin. Oncol. 2011, 2, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, Z.B.; Chen, W.Z.; Bai, J.; Zhu, H.; Qiao, T.Y. Preliminary Experience Using High Intensity Focused Ultrasound for the Treatment of Patients with Advanced Stage Renal Malignancy. J. Urol. 2003, 170, 2237–2240. [Google Scholar] [CrossRef] [Green Version]
- Berge, V.; Dickinson, L.; McCartan, N.; Hindley, R.G.; Diep, L.M.; Emberton, M.; Ahmed, H.U. Morbidity Associated with Primary High Intensity Focused Ultrasound and Redo High Intensity Focused Ultrasound for Localized Prostate Cancer. J. Urol. 2014, 191, 1764–1769. [Google Scholar] [CrossRef]
- Miller, D.L.; Smith, N.B.; Bailey, M.R.; Czarnota, G.J.; Hynynen, K.; Makin, I.R.S. Overview of Therapeutic Ultrasound Applications and Safety Considerations. J. Ultras Med. 2012, 31, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Tavakolinejad, A.; Rabbani, M.; Janmaleki, M. Effects of Hypergravity on Adipose-Derived Stem Cell Morphology, Mechanical Property and Proliferation. Biochem. Biophys. Res. Commun. 2015, 464, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M.; Yoshida, S. Stimulation of DNA Polymerase α by Hypergravity Generated by Centrifugal Acceleration. Biochem. Biophys. Res. Commun. 2001, 289, 345–349. [Google Scholar] [CrossRef]
- Thiel, C.S.; Tauber, S.; Christoffel, S.; Huge, A.; Lauber, B.A.; Polzer, J.; Paulsen, K.; Lier, H.; Engelmann, F.; Schmitz, B.; et al. Rapid Coupling between Gravitational Forces and the Transcriptome in Human Myelomonocytic U937 Cells. Sci. Rep. 2018, 8, 1–24. [Google Scholar]
- Thiel, C.S.; Hauschild, S.; Tauber, S.; Paulsen, K.; Raig, C.; Raem, A.; Biskup, J.; Gutewort, A.; Hürlimann, E.; Unverdorben, F.; et al. Identification of Reference Genes in Human Myelomonocytic Cells for Gene Expression Studies in Altered Gravity. BioMed Res. Int. 2015, 2015, 363575. [Google Scholar] [CrossRef]
- Costa-Almeida, R.; Carvalho, D.T.O.; Ferreira, M.J.S.; Pesqueira, T.; Monici, M.; van Loon, J.J.W.A.; Granja, P.L.; Gomes, M.E. Continuous Exposure to Simulated Hypergravity-Induced Changes in Proliferation, Morphology, and Gene Expression of Human Tendon Cells. Stem Cells Dev. 2018, 27, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Hemmersbach, R.; von der Wiesche, M.; Seibt, D. Ground-Based Experimental Platforms in Gravitational Biology and Human Physiology. Signal Transduct. 2006, 6, 381–387. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Guo, S.; Li, B.-B.; Jiang, N.; Li, A.; Yan, H.-F.; Yang, H.-M.; Zhou, J.-L.; Li, C.-L.; Cui, Y. Effect of Weightlessness on the 3D Structure Formation and Physiologic Function of Human Cancer Cells. BioMed Res. Int. 2019, 2019, 4894083. [Google Scholar] [CrossRef] [Green Version]
- Frett, T.; Petrat, G.; van Loon, J.J.; Hemmersbach, R.; Anken, R. Hypergravity Facilities in the ESA Ground-Based Facility Program− Current Research Activities and Future Tasks. Microgravity Sci. Technol. 2016, 28, 205–214. [Google Scholar] [CrossRef]
- van Loon, J.J.W.A. The Human Centrifuge. Microgravity Sci. Technol. 2009, 21, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Kopp, S.; Krüger, M.; Feldmann, S.; Oltmann, H.; Schütte, A.; Schmitz, B.; Bauer, J.; Schulz, H.; Saar, K.; Huebner, N.; et al. Thyroid Cancer Cells in Space during the TEXUS-53 Sounding Rocket Mission-The THYROID Project OPEN. Sci. Rep. 2018, 8, 10355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehland, M.; Aleshcheva, G.; Schulz, H.; Saar, K.; Hübner, N.; Hemmersbach, R.; Braun, M.; Ma, X.; Frett, T.; Warnke, E.; et al. Differential Gene Expression of Human Chondrocytes Cultured under Short-Term Altered Gravity Conditions during Parabolic Flight Maneuvers. Cell Commun. Signal. 2015, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Guo, Y.B.; Zhang, M.; Sun, Y.Q. The Subsequent Biological Effects of Simulated Microgravity on Endothelial Cell Growth in HUVECs. Chin. J. Traumatol. 2018, 21, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, A.; Cogoli, A. Hypergravity Promotes Cell Proliferation. Experientia 1983, 39, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Cao, Y.; Zhang, Y.; Qian, J.; Zhao, Q.; Liu, F.; Zhang, T.; Zhou, J.; Gu, Y.; Xia, G.; et al. Effect of Microgravity on Proliferation and Differentiation of Embryonic Stem Cells in an Automated Culturing System during the TZ-1 Space Mission. Cell Prolif. 2018, 51, e12466. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jeong, A.J.; Kim, M.; Lee, C.; Ye, S.K.; Kim, S. Time-Averaged Simulated Microgravity (TaSMG) Inhibits Proliferation of Lymphoma Cells, L-540 and HDLM-2, Using a 3D Clinostat. Biomed. Eng. Online 2017, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sokolovskaya, A.A.; Ignashkova, T.I.; Bochenkova, A.V.; Moskovtsev, A.A.; Baranov, V.M.; Kubatiev, A.A. Effects of Simulated Microgravity on Cell Cycle in Human Endothelial Cells. Acta Astronaut. 2014, 99, 16–23. [Google Scholar] [CrossRef]
- Arun, R.P.; Sivanesan, D.; Vidyasekar, P.; Verma, R.S. PTEN/FOXO3/AKT Pathway Regulates Cell Death and Mediates Morphogenetic Differentiation of Colorectal Cancer Cells under Simulated Microgravity. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rocca, A.; Marino, A.; Rocca, V.; Moscato, S.; de Vito, G.; Piazza, V.; Mazzolai, B.; Mattoli, V.; Ngo-Anh, T.J.; Ciofani, G. Barium Titanate Nanoparticles and Hypergravity Stimulation Improve Differentiation of Mesenchymal Stem Cells into Osteoblasts. Int. J. Nanomed. 2015, 10, 433–445. [Google Scholar]
- Montufar-Solis, D.; Duke, P.; D’Aunno, D. In Vivo and in Vitro Studies of Cartilage Differentiation in Altered Gravities. Adv. Sp. Res. 1996, 17, 193–199. [Google Scholar] [CrossRef]
- Li, C.F.; Sun, J.X.; Gao, Y.; Shi, F.; Pan, Y.K.; Wang, Y.C.; Sun, X.Q. Clinorotation-Induced Autophagy via HDM2-P53-MTOR Pathway Enhances Cell Migration in Vascular Endothelial Cells. Cell Death Dis. 2018, 9, 147. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Lu, D.-Y.; Shi, F.; Zhang, S.; Yang, C.-B.; Wang, B.; Cao, X.-S.; Du, T.-Y.; Gao, Y.; Zhao, J.-D.; et al. Clinorotation Enhances Autophagy in Vascular Endothelial Cells. Biochem. Cell. Biol. Biol. Cell. 2013, 91, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.Y.; Zou, L.; Yuan, M.; Wang, Y.; Li, T.Z.; Zhang, Y.; Wang, J.F.; Li, Y.; Deng, X.W.; Liu, C.T. Impact of Simulated Microgravity on Microvascular Endothelial Cell Apoptosis. Eur. J. Appl. Physiol. 2011, 111, 2131–2138. [Google Scholar] [CrossRef]
- Sundaresan, A.; Risin, D.; Pellis, N.R. Cell growth in microgravity. In Encyclopedia of Molecular Cell Biology and Molecular Medicine; Meyers, R.A., Sendtko, A., Henheik, P., Eds.; Wiley-VCH: Weinheim, Germany, 2004; Volume 2, pp. 303–321. [Google Scholar]
- Lambert, C.A.; Lapière, C.M.; Nusgens, B.V. Biology of adherent cells in microgravity. In Biology in Space and Life on Earth; Brinckmann, E., Ed.; Wiley-VCH: New York, NY, USA, 2007; pp. 123–155. [Google Scholar]
- Rudimov, E.G.; Buravkova, L.B. Gravisensitivity of Endothelial Cells: The Role of Cytoskeleton and Adhesion Molecules. Hum. Physiol. 2016, 42, 687–693. [Google Scholar] [CrossRef]
- Croute, F.; Gaubin, Y.; Pianezzi, B.; Soleilhavoup, J.P. Effects of Hypergravity on the Cell Shape and on the Organization of Cytoskeleton and Extracelluar Matrix Molecules of in Vitro Human Dermal Fibroblasts. Microgravity Sci. Technol. 1995, 8, 118–124. [Google Scholar] [PubMed]
- Kelly, D.J.; Jacobs, C.R. The Role of Mechanical Signals in Regulating Chondrogenesis and Osteogenesis of Mesenchymal Stem Cells. Birth Defects Res. Part C 2010, 90, 75–85. [Google Scholar] [CrossRef]
- Bacso, Z.; Nagy, H.; Goda, K.; Bene, L.; Fenyvesi, F.; Matkó, J.; Szabó, G. Raft and Cytoskeleton Associations of an ABC Transporter: P-Glycoprotein. Cytom. Part A 2004, 61, 105–116. [Google Scholar] [CrossRef]
- Albi, E.; Curcio, F.; Lazzarini, A.; Floridi, A.; Cataldi, S.; Lazzarini, R.; Loreti, E.; Ferri, I.; Ambesi-Impiombato, F.S. A Firmer Understanding of the Effect of Hypergravity on Thyroid Tissue: Cholesterol and Thyrotropin Receptor. PLoS ONE 2014, 9, 5–11. [Google Scholar] [CrossRef]
- Zhou, S.; Zu, Y.; Zhuang, F.; Yang, C. Hypergravity-Induced Enrichment of Β1 Integrin on the Cell Membranes of Osteoblast-like Cells via Caveolae-Dependent Endocytosis. Biochem. Biophys. Res. Commun. 2015, 463, 928–933. [Google Scholar] [CrossRef]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and Cancer: Regulators of Cancer Stemness, Metastasis, and Drug Resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Janmaleki, M.; Pachenari, M.; Seyedpour, S.M.; Shahghadami, R.; Sanati-Nezhad, A. Impact of Simulated Microgravity on Cytoskeleton and Viscoelastic Properties of Endothelial Cell. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuimova, M.K.; Yahioglu, G.; Ogilby, P.R. Singlet Oxygen in a Cell: Spatially Dependent Lifetimes and Quenching Rate Constants. J. Am. Chem. Soc. 2009, 131, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.; Choquet, D.; Triller, A. Control of the Postsynaptic Membrane Viscosity. J. Neurosci. 2009, 29, 2926–2937. [Google Scholar] [CrossRef] [PubMed]
- Kalwarczyk, T.; Ziȩbacz, N.; Bielejewska, A.; Zaboklicka, E.; Koynov, K.; Szymański, J.; Wilk, A.; Patkowski, A.; Gapiński, J.; Butt, H.-J.; et al. Comparative Analysis of Viscosity of Complex Liquids and Cytoplasm of Mammalian Cells at the Nanoscale. Nano Lett. 2011, 11, 2157–2163. [Google Scholar] [CrossRef]
- Woodcock, E.M.; Girvan, P.; Eckert, J.; Lopez-Duarte, I.; Kubánková, M.; van Loon, J.J.W.A.; Brooks, N.J.; Kuimova, M.K. Measuring Intracellular Viscosity in Conditions of Hypergravity. Biophys. J. 2019, 116, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Kohn, F.P.M.; Hauslage, J. The Gravity Dependence of Pharmacodynamics: The Integration of Lidocaine into Membranes in Microgravity. Npj Microgravity 2019, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Sieber, M.; Hanke, W.; Kohn, F.P.M. Modification of Membrane Fluidity by Gravity. Open J. Biophys. 2014, 4, 105–111. [Google Scholar] [CrossRef] [Green Version]




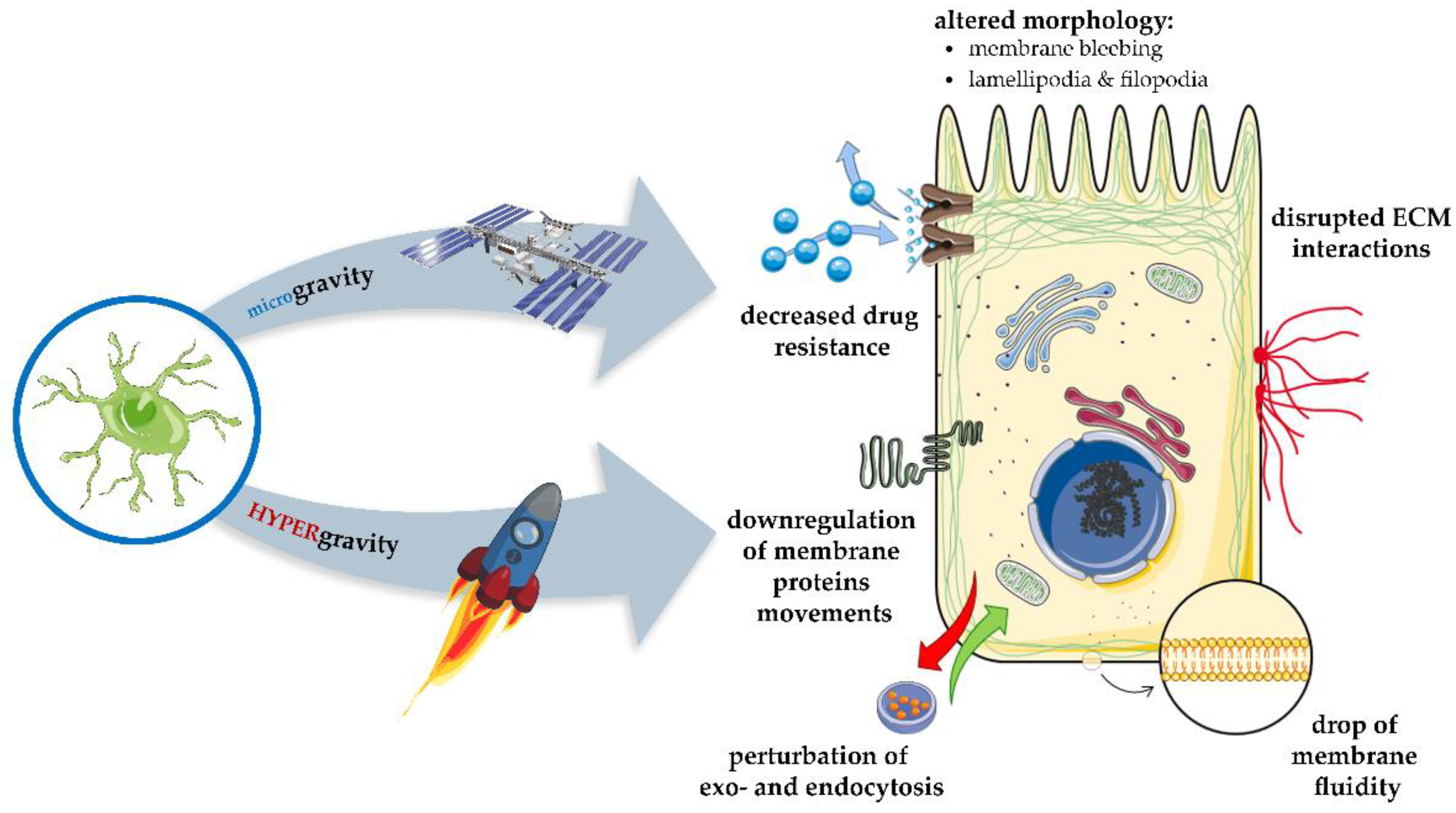

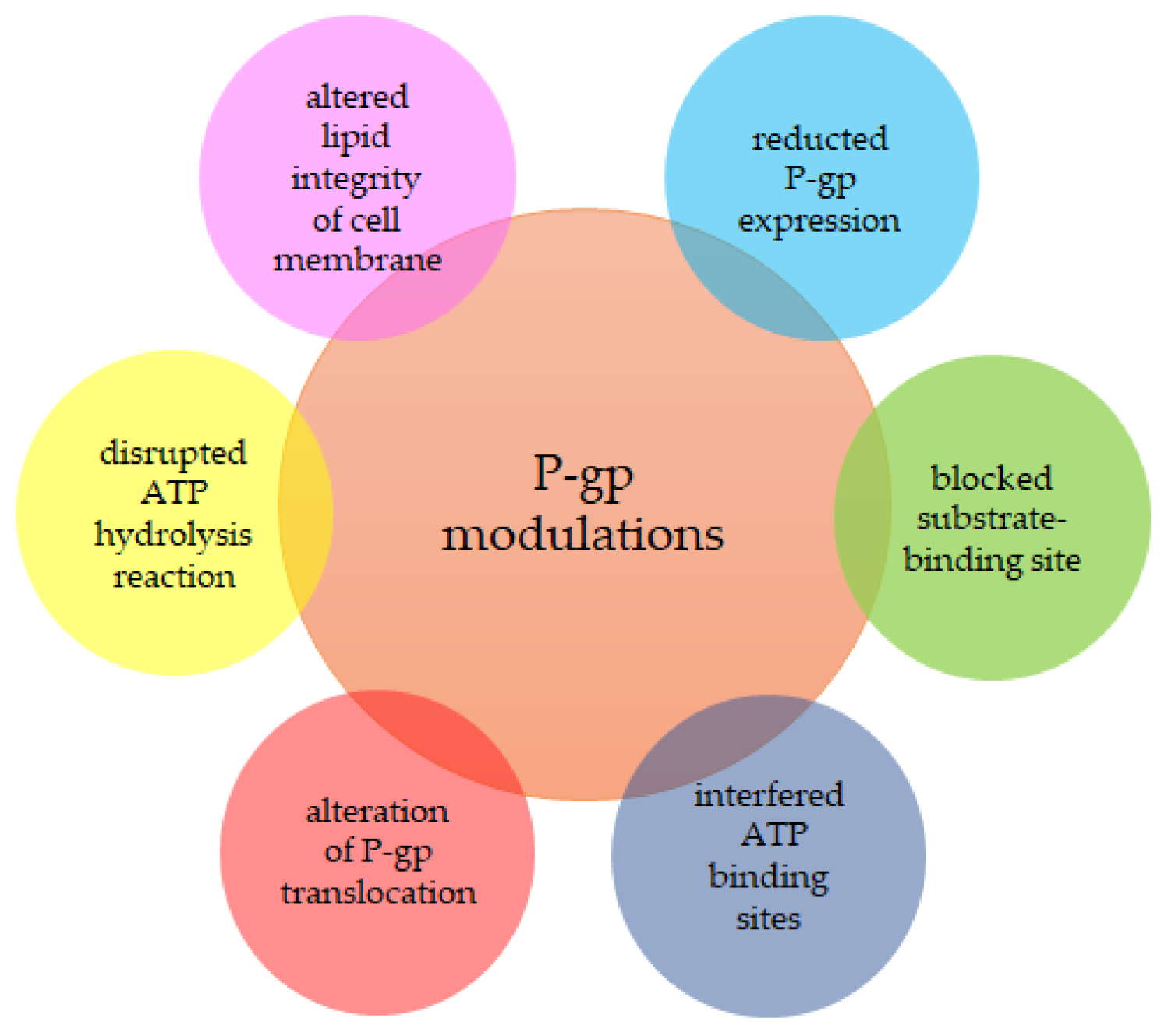{kind=link}
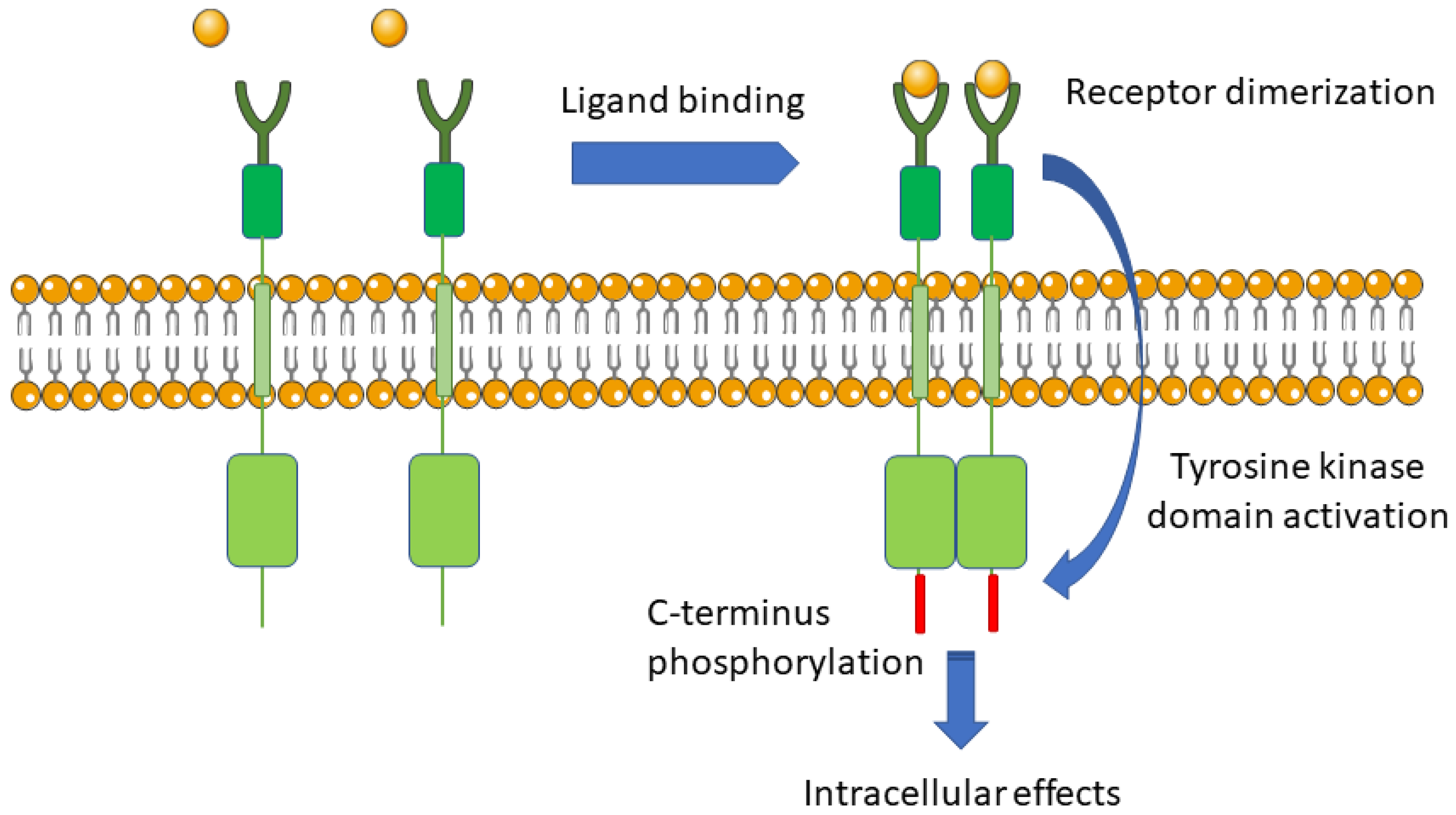{kind=link}
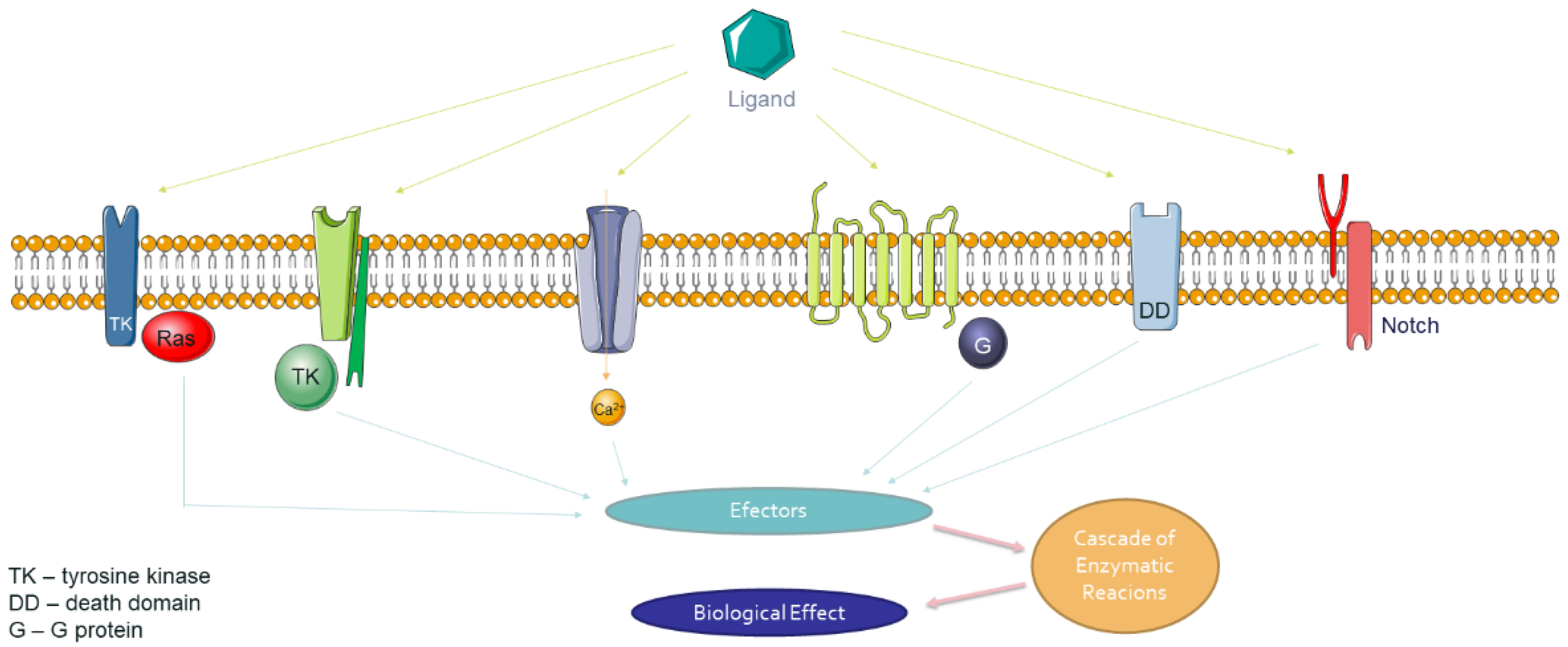{kind=link}
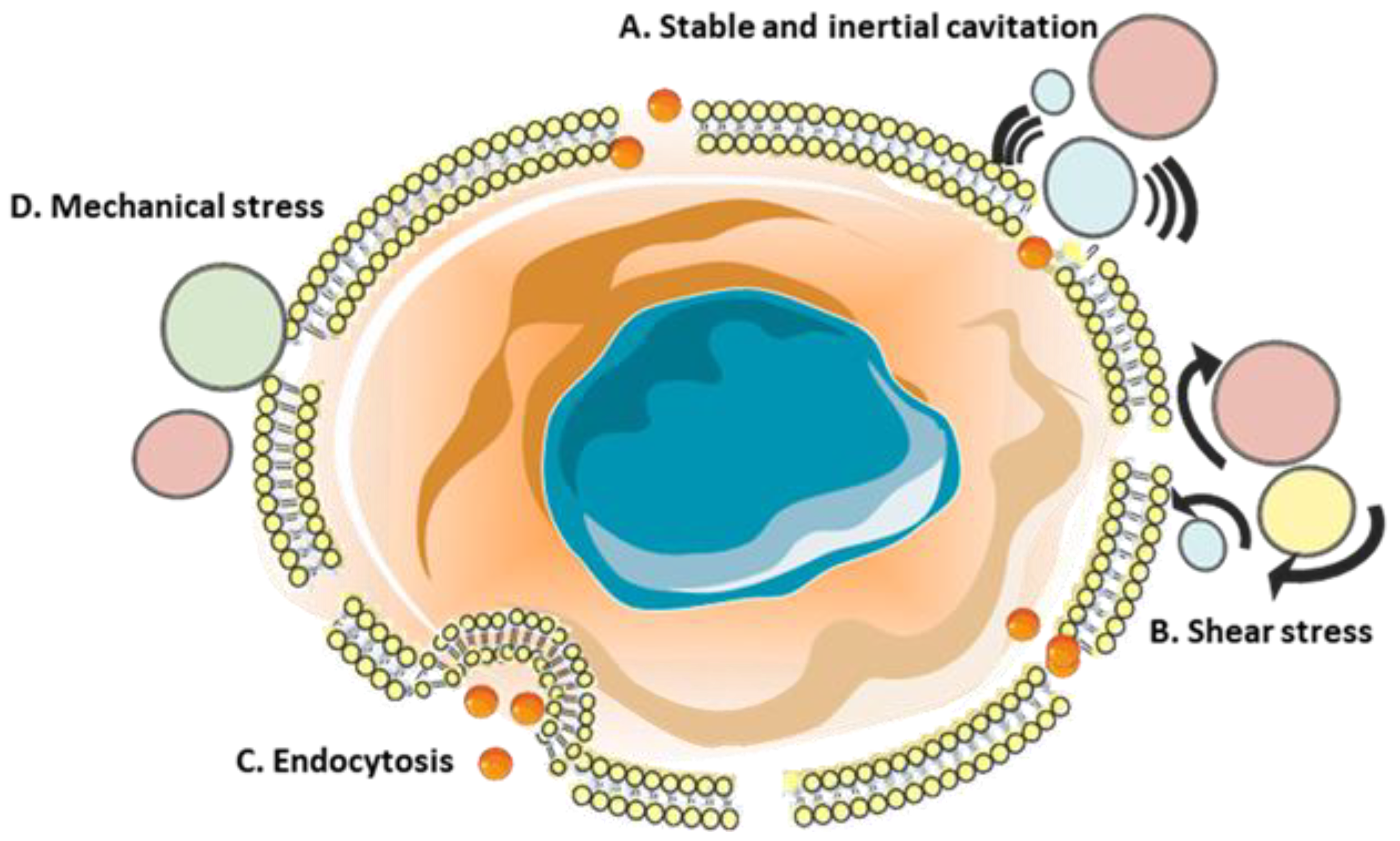{kind=link}
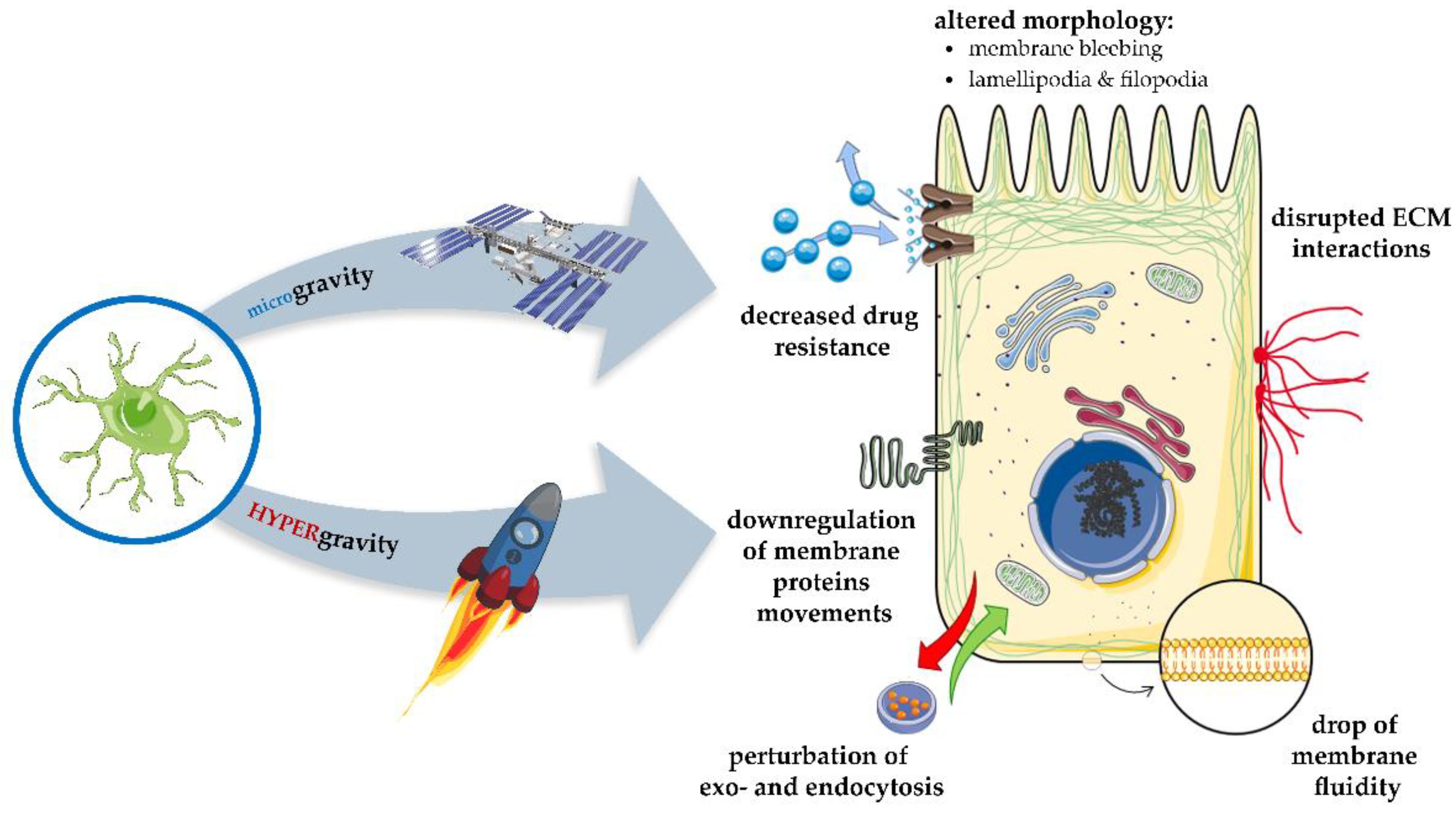{kind=link}
| Method | Type of Membrane Modification | Expected Outcomes | Literature |
|---|---|---|---|
| MDR modulators | Translocation of the ABC superfamily proteins Decrease in the level of ABC superfamily proteins Altered lipid integrity, fluidity or permeability of cell membrane Modulation of ABC superfamily proteins activity | Increase in cellular sensitivity to anti-cancer drugs/decrease of MDR | [35] [48] [52,54,55] [31,34,41,47,49,50,51,52,53,56,57,58,59,60,61,62,63,64] |
| MicroRNAs as regulators of MDR | Decrease in the level of ABC superfamily proteins | Increase in cellular sensitivity to anti-cancer drugs/decrease of MDR | [74,75,76,77,78,79,80,81,82,83,84,85,86,87] |
| Modification of IC | Diminish of IC activity by natural and synthetic inhibitors IC inhibition by antibodies IC expression decrease by siRNA, miRNA, CRISPR/Cas9 | Alterations in ion efflux/influx; inhibition of cell proliferation, motility, and invasiveness; increase of cell apoptosis and sensitivity for anticancer drugs | [106,111,113,116,122] [127,128] [132,133,134,135] |
| Membrane receptors modulations | Inhibition of membrane receptors | Sensitize cancer cells to conventional therapy | [155,156,157,158,159,160,164,165,166,167,168,169] |
| Membrane lipidomics modulations | Changes in the composition of membrane lipids Changes in the activity of membrane-related enzymes and signaling pathways | Increased membrane permeability, decreased drug resistance Sensitize cancer cells to conventional therapy | [3,171,172,173,175,176,178,179,181,182,185,186] [177,186] |
| Electroporation | Induction of pores in the lipid membrane. Irreversible pores-membrane disruption | Increased membrane permeability/cell lysis, delivering drugs into the cell | [188,189,190,191,192,195,202,203,204,205,206,220,221,222,223,224,225,229,230,231,233,234,235,236,238,239,242,243] |
| Sonoporation | Membrane invaginations—pores, endocytotic vesicles; membrane disruption | Increased membrane permeability/cell lysis, delivering drugs into the cell | [252,253,268,270] |
| Gravitational forces | Membrane blebbing; drop of membrane fluidity; disrupted ECM interactions and membrane proteins movements | Decreased drug resistance; altered cell morphology | [294,295,296,297,298,299] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choromańska, A.; Chwiłkowska, A.; Kulbacka, J.; Baczyńska, D.; Rembiałkowska, N.; Szewczyk, A.; Michel, O.; Gajewska-Naryniecka, A.; Przystupski, D.; Saczko, J. Modifications of Plasma Membrane Organization in Cancer Cells for Targeted Therapy. Molecules 2021, 26, 1850. https://doi.org/10.3390/molecules26071850
Choromańska A, Chwiłkowska A, Kulbacka J, Baczyńska D, Rembiałkowska N, Szewczyk A, Michel O, Gajewska-Naryniecka A, Przystupski D, Saczko J. Modifications of Plasma Membrane Organization in Cancer Cells for Targeted Therapy. Molecules. 2021; 26(7):1850. https://doi.org/10.3390/molecules26071850
Chicago/Turabian StyleChoromańska, Anna, Agnieszka Chwiłkowska, Julita Kulbacka, Dagmara Baczyńska, Nina Rembiałkowska, Anna Szewczyk, Olga Michel, Agnieszka Gajewska-Naryniecka, Dawid Przystupski, and Jolanta Saczko. 2021. "Modifications of Plasma Membrane Organization in Cancer Cells for Targeted Therapy" Molecules 26, no. 7: 1850. https://doi.org/10.3390/molecules26071850
APA StyleChoromańska, A., Chwiłkowska, A., Kulbacka, J., Baczyńska, D., Rembiałkowska, N., Szewczyk, A., Michel, O., Gajewska-Naryniecka, A., Przystupski, D., & Saczko, J. (2021). Modifications of Plasma Membrane Organization in Cancer Cells for Targeted Therapy. Molecules, 26(7), 1850. https://doi.org/10.3390/molecules26071850