
Posttranslational Chemical Mutagenesis Methods to Insert Posttranslational Modifications into Recombinant Proteins



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
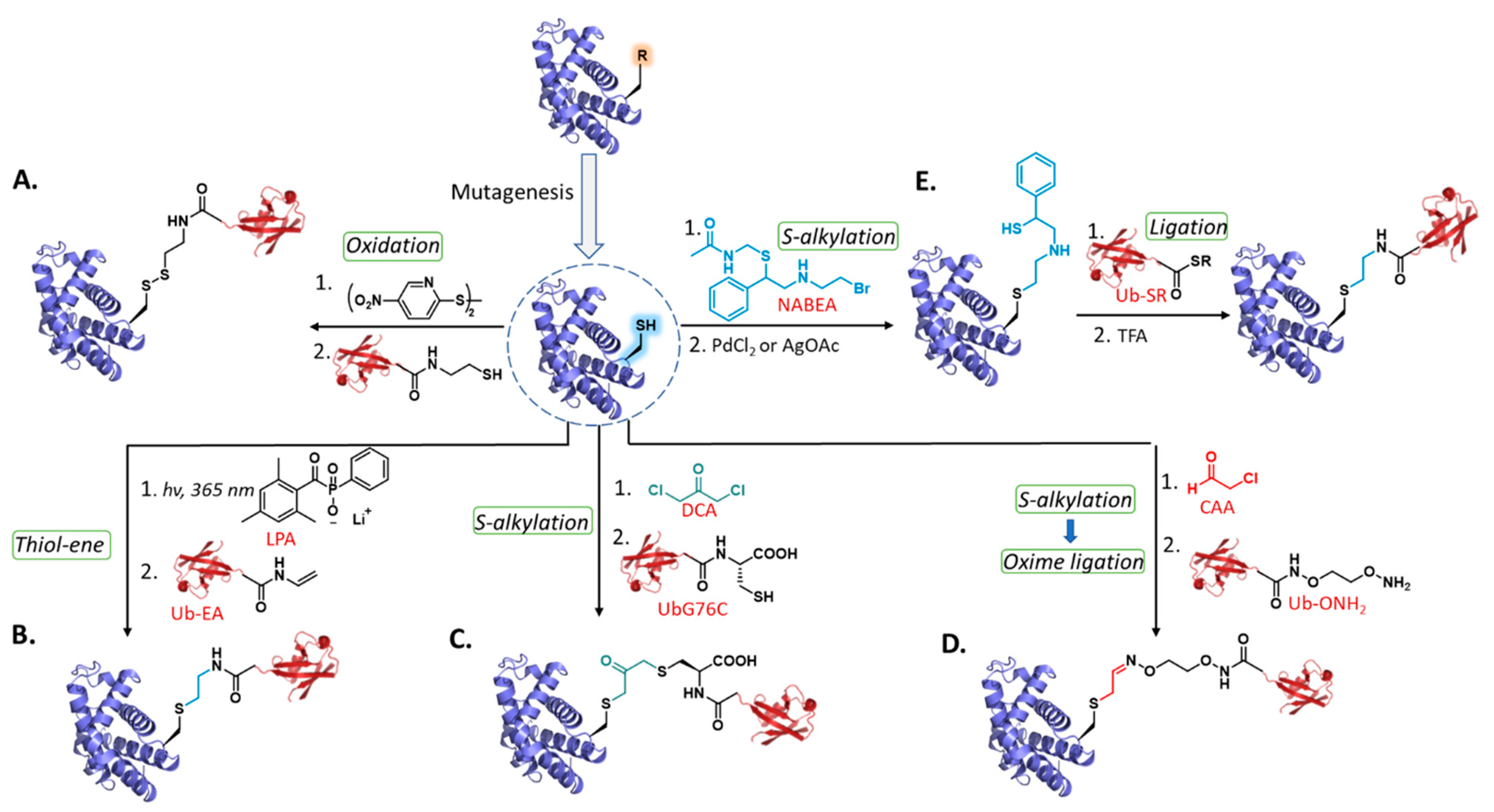
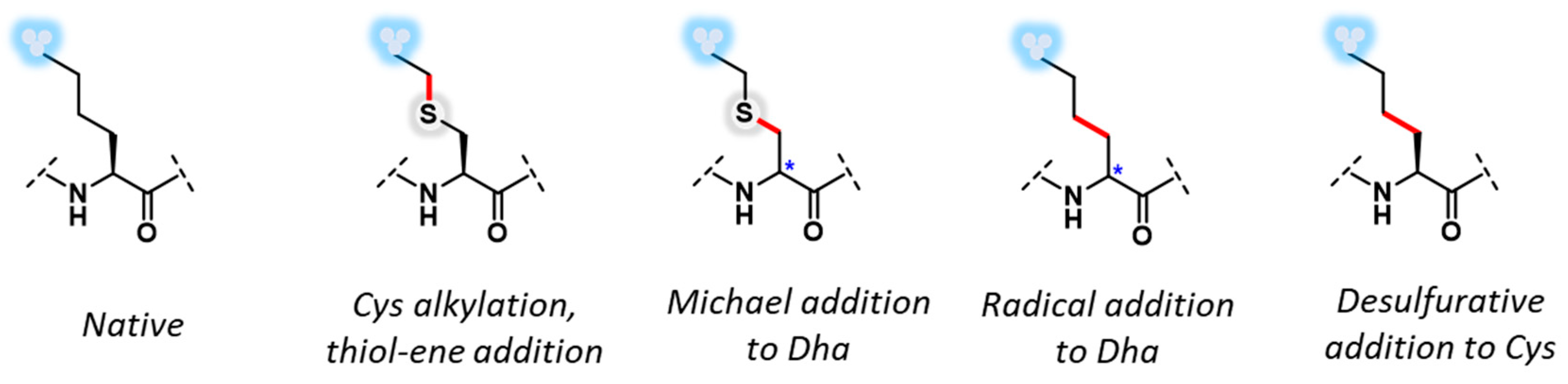
2. Late-Stage Cys Functionalization
3. Dehydroalanine Coupled via Michael Addition Chemistry
4. Carbon–Carbon Bond-Forming Reactions
5. Conclusions and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C. Deciphering Protein Post-Translational Modifications Using Chemical Biology Tools. Nat. Rev. Chem. 2020, 4, 674–695. [Google Scholar] [CrossRef]
- Krall, N.; Da Cruz, F.P.; Boutureira, O.; Bernardes, G.J.L. Site-Selective Protein-Modification Chemistry for Basic Biology and Drug Development. Nat. Chem. 2016, 8, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Spicer, C.D.; Davis, B.G. Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 4740. [Google Scholar] [CrossRef] [Green Version]
- Lateef, O.M.; Akintubosun, M.O.; Olaoba, O.T.; Samson, S.O.; Adamczyk, M. Making Sense of “Nonsense” and More: Challenges and Opportunities in the Genetic Code Expansion, in the World of TRNA Modifications. Int. J. Mol. Sci. 2022, 23, 93. [Google Scholar]
- Xie, J.; Schultz, P.G. A Chemical Toolkit for Proteins—An Expanded Genetic Code. Nat. Rev. Mol. Cell Biol. 2006, 7, 775–782. [Google Scholar] [CrossRef]
- Bondalapati, S.; Jbara, M.; Brik, A. Expanding the Chemical Toolbox for the Synthesis of Large and Uniquely Modified Proteins. Nat. Chem. 2016, 8, 407–418. [Google Scholar]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoët, M.; Monbaliu, J.C.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Sayers, J.; Premdjee, B.; Payne, R.J. Rapid and Efficient Protein Synthesis through Expansion of the Native Chemical Ligation Concept. Nat. Rev. Chem. 2018, 2, 122. [Google Scholar] [CrossRef]
- Maity, S.K.; Jbara, M.; Brik, A. Chemical and Semisynthesis of Modified Histones. J. Pept. Sci. 2016, 22, 252–259. [Google Scholar] [CrossRef]
- Conibear, A.C.; Watson, E.E.; Payne, R.J.; Becker, C.F.W. Native Chemical Ligation in Protein Synthesis and Semi-Synthesis. Chem. Soc. Rev. 2018, 47, 9046–9068. [Google Scholar] [CrossRef] [PubMed]
- Hackenberger, C.P.R.; Schwarzer, D. Chemoselective Ligation and Modification Strategies for Peptides and Proteins. Angew. Chem. Int. Ed. 2008, 47, 10030–10074. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.W.; Fox, R.M.; Baucom, K.D. Chemoselective Amide Ligations by Decarboxylative Condensations of N-Alkylhydroxylamines and α-Ketoacids. Angew. Chem. Int. Ed. 2006, 118, 1270–1274. [Google Scholar] [CrossRef]
- Mitchell, N.J.; Malins, L.R.; Liu, X.; Thompson, R.E.; Chan, B.; Radom, L.; Payne, R.J. Rapid Additive-Free Selenocystine-Selenoester Peptide Ligation. J. Am. Chem. Soc. 2015, 137, 14011–14014. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, C.; Lam, H.Y.; Lee, C.L.; Li, X. Protein Chemical Synthesis by Serine and Threonine Ligation. Proc. Natl. Acad. Sci. USA 2013, 110, 6657–6662. [Google Scholar] [CrossRef] [Green Version]
- Jbara, M.; Maity, S.K.; Seenaiah, M.; Brik, A. Palladium Mediated Rapid Deprotection of N-Terminal Cysteine under Native Chemical Ligation Conditions for the Efficient Preparation of Synthetically Challenging Proteins. J. Am. Chem. Soc. 2016, 138, 5069–5075. [Google Scholar] [CrossRef]
- Kamo, N.; Hayashi, G.; Okamoto, A. Triple Function of 4-Mercaptophenylacetic Acid Promotes One-Pot Multiple Peptide Ligation. Angew. Chem. Int. Ed. 2018, 57, 16533–16537. [Google Scholar] [CrossRef]
- Jbara, M.; Eid, E.; Brik, A. Gold(I)-Mediated Decaging or Cleavage of Propargylated Peptide Bond in Aqueous Conditions for Protein Synthesis and Manipulation. J. Am. Chem. Soc. 2020, 142, 8203–8210. [Google Scholar] [CrossRef]
- Li, J.B.; Tang, S.; Zheng, J.S.; Tian, C.L.; Liu, L. Removable Backbone Modification Method for the Chemical Synthesis of Membrane Proteins. Acc. Chem. Res. 2017, 50, 1143–1153. [Google Scholar] [CrossRef]
- Hartrampf, N.; Saebi, A.; Poskus, M.; Gates, Z.P.; Callahan, A.J.; Cowfer, A.E.; Hanna, S.; Antilla, S.; Schissel, C.K.; Quartararo, A.J.; et al. Synthesis of Proteins by Automated Flow Chemistry. Science 2020, 368, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Gates, Z.P.; Hartrampf, N. Flow-Based SPPS for Protein Synthesis: A Perspective. Pept. Sci. 2020, 112, e24198. [Google Scholar] [CrossRef]
- Durek, T.; Becker, C.F.W. Protein Semi-Synthesis: New Proteins for Functional and Structural Studies. Biomol. Eng. 2005, 22, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.; Muir, T. Application of the Protein Semisynthesis Strategy to the Generation of Modified Chromatin. Annu. Rev. Biochem. 2015, 84, 265. [Google Scholar] [CrossRef] [Green Version]
- Jbara, M.; Maity, S.K.; Brik, A. Examining Several Strategies for the Chemical Synthesis of Phosphorylated Histone H3 Reveals the Effectiveness of the Convergent Approach. Eur. J. Org. Chem. 2019, 21, 3128–3132. [Google Scholar] [CrossRef]
- Haj-Yahya, M.; Lashuel, H.A. Protein Semisynthesis Provides Access to Tau Disease-Associated Post-Translational Modifications (PTMs) and Paves the Way to Deciphering the Tau PTM Code in Health and Diseased States. J. Am. Chem. Soc. 2018, 140, 6611–6621. [Google Scholar] [CrossRef] [Green Version]
- Kamo, N.; Hayashi, G.; Okamoto, A. Chemical Synthesis of Cys-Containing Protein via Chemoselective Deprotection with Different Palladium Complexes. Org. Lett. 2019, 21, 8378–8382. [Google Scholar] [CrossRef]
- Lechner, C.C.; Agashe, N.D.; Fierz, B. Traceless Synthesis of Asymmetrically Modified Bivalent Nucleosomes. Angew. Chem. Int. Ed. 2016, 55, 2903–2906. [Google Scholar] [CrossRef]
- Seenaiah, M.; Jbara, M.; Mali, S.M.; Brik, A. Convergent Versus Sequential Protein Synthesis: The Case of Ubiquitinated and Glycosylated H2B. Angew. Chem. Int. Ed. 2015, 54, 12374–12378. [Google Scholar] [CrossRef]
- Maki, Y.; Okamoto, R.; Izumi, M.; Kajihara, Y. Chemical Synthesis of an Erythropoietin Glycoform Having a Triantennary N-Glycan: Significant Change of Biological Activity of Glycoprotein by Addition of a Small Molecular Weight Trisaccharide. J. Am. Chem. Soc. 2020, 142, 20671–20679. [Google Scholar] [CrossRef]
- Jbara, M.; Guttmann-Raviv, N.; Maity, S.K.; Ayoub, N.; Brik, A. Total Chemical Synthesis of Methylated Analogues of Histone 3 Revealed KDM4D as a Potential Regulator of H3K79me3. Bioorganic Med. Chem. 2017, 25, 4966–4970. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.K.; He, Q.Q.; Ai, H.S.; Guo, J.; Li, J. The Convergent Chemical Synthesis of Histone H3 Protein for Site-Specific Acetylation at Lys56 and Ubiquitination at Lys122. Chem. Commun. 2017, 53, 4148–4151. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.E.; Liu, X.; Thompson, R.E.; Ripoll-Rozada, J.; Wu, M.; Alwis, I.; Gori, A.; Loh, C.T.; Parker, B.L.; Otting, G.; et al. Mosquito-Derived Anophelin Sulfoproteins Are Potent Antithrombotics. ACS Cent. Sci. 2018, 4, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Clayton, D.; Kulkarni, S.S.; Sayers, J.; Dowman, L.J.; Ripoll-Rozada, J.; Pereira, P.J.B.; Payne, R.J. Chemical Synthesis of a Haemathrin Sulfoprotein Library Reveals Enhanced Thrombin Inhibition Following Tyrosine Sulfation. RSC Chem. Biol. 2020, 1, 379–384. [Google Scholar] [CrossRef]
- Bouchenna, J.; Sénéchal, M.; Drobecq, H.; Vicogne, J.; Melnyk, O. Total Chemical Synthesis of All SUMO-2/3 Dimer Combinations. Bioconjug. Chem. 2019, 30, 2967–2973. [Google Scholar] [CrossRef] [PubMed]
- Weller, C.E.; Dhall, A.; Ding, F.; Linares, E.; Whedon, S.D.; Senger, N.A.; Tyson, E.L.; Bagert, J.D.; Li, X.; Augusto, O.; et al. Aromatic Thiol-Mediated Cleavage of N-O Bonds Enables Chemical Ubiquitylation of Folded Proteins. Nat. Commun. 2016, 7, 2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinty, R.K.; Kim, J.; Chatterjee, C.; Roeder, R.G.; Muir, T.W. Chemically Ubiquitylated Histone H2B Stimulates HDot1L-Mediated Intranucleosomal Methylation. Nature 2008, 453, 812–816. [Google Scholar] [CrossRef]
- Nune, M.; Morgan, M.T.; Connell, Z.; McCullough, L.; Jbara, M.; Sun, H.; Brik, A.; Formosa, T.; Wolberger, C. FACT and Ubp10 Collaborate to Modulate H2B Deubiquitination and Nucleosome Dynamics. eLife 2019, 8, e40988. [Google Scholar] [CrossRef]
- Tan, Y.; Wu, H.; Wei, T.; Li, X. Chemical Protein Synthesis: Advances, Challenges, and Outlooks. J. Am. Chem. Soc. 2020, 142, 20288–20298. [Google Scholar] [CrossRef]
- Boutureira, O.; Bernardes, G.J.L. Advances in Chemical Protein Modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef]
- Jbara, M.; Maity, S.K.; Brik, A. Palladium in the Chemical Synthesis and Modification of Proteins. Angew. Chem. Int. Ed. 2017, 56, 10644–10655. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.M.; Bernardes, G.J.L.; Lin, Y.A.; Davis, B.G. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Jbara, M. Transition Metal Catalyzed Site-Selective Cysteine Diversification of Proteins. Pure Appl. Chem. 2021, 93, 169–186. [Google Scholar] [CrossRef]
- Bernardes, G.J.L.; Chalker, J.M.; Errey, J.C.; Davis, B.G. Facile Conversion of Cysteine and Alkyl Cysteines to Dehydroalanine on Protein Surfaces: Versatile and Switchable Access to Functionalized Proteins. J. Am. Chem. Soc. 2008, 130, 5052–5053. [Google Scholar] [CrossRef] [PubMed]
- Jbara, M.; Laps, S.; Morgan, M.; Kamnesky, G.; Mann, G.; Wolberger, C.; Brik, A. Palladium Prompted On-Demand Cysteine Chemistry for the Synthesis of Challenging and Uniquely Modified Proteins. Nat. Commun. 2018, 9, 3154. [Google Scholar] [CrossRef] [Green Version]
- Macmillan, D.; Bill, R.M.; Sage, K.A.; Fern, D.; Flitsch, S.L. Selective in Vitro Glycosylation of Recombinant Proteins: Semi-Synthesis of Novel Homogeneous Glycoforms of Human Erythropoietin. Chem. Biol. 2001, 8, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.D.; Chu, F.; Racki, L.R.; de la Cruz, C.C.; Burlingame, A.L.; Panning, B.; Narlikar, G.J.; Shokat, K.M. The Site-Specific Installation of Methyl-Lysine Analogs into Recombinant Histones. Cell 2007, 128, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Jbara, M.; Pomplun, S.; Schissel, C.K.; Hawken, S.W.; Boija, A.; Klein, I.; Rodriguez, J.; Buchwald, S.L.; Pentelute, B.L. Engineering Bioactive Dimeric Transcription Factor Analogs via Palladium Rebound Reagents. J. Am. Chem. Soc. 2021, 143, 11788–11798. [Google Scholar] [CrossRef]
- Jbara, M.; Rodriguez, J.; Dhanjee, H.H.; Loas, A.; Buchwald, S.L.; Pentelute, B.L. Oligonucleotide Bioconjugation with Bifunctional Palladium Reagents. Angew. Chem. Int. Ed. 2021, 60, 12109–12115. [Google Scholar] [CrossRef]
- Gunnoo, S.B.; Madder, A. Chemical Protein Modification through Cysteine. ChemBioChem 2016, 17, 529–553. [Google Scholar] [CrossRef] [Green Version]
- Dadová, J.; Galan, S.R.; Davis, B.G. Synthesis of Modified Proteins via Functionalization of Dehydroalanine. Curr. Opin. Chem. Biol. 2018, 46, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.H.; Vallée, M.R.J.; Davis, B.G. From Chemical Mutagenesis to Post-Expression Mutagenesis: A 50 Year Odyssey. Angew. Chem. Int. Ed. 2016, 50, 5896–5903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephanopoulos, N.; Francis, M.B. Choosing an Effective Protein Bioconjugation Strategy. Nat. Chem. Biol. 2011, 7, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Ochtrop, P.; Hackenberger, C.P.R. Recent Advances of Thiol-Selective Bioconjugation Reactions. Curr. Opin. Chem. Biol. 2020, 58, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, Z.; Tian, G.; Bao, X.; Ishibashi, T.; Li, X.D. Site-Specific Installation of Succinyl Lysine Analog into Histones Reveals the Effect of H2BK34 Succinylation on Nucleosome Dynamics. Cell Chem. Biol. 2018, 25, 166–174.e7. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Allahverdi, A.; Yang, R.; Lua, G.B.J.; Zhang, X.; Cao, Y.; Korolev, N.; Nordenskiöld, L.; Liu, C.F. A Direct Method for Site-Specific Protein Acetylation. Angew. Chem. Int. Ed. 2011, 50, 9611–9614. [Google Scholar] [CrossRef]
- Gui, W.; Davidson, G.A.; Zhuang, Z. Chemical Methods for Protein Site-Specific Ubiquitination. RSC Chem. Biol. 2021, 2, 450–467. [Google Scholar] [CrossRef]
- Huang, R.; Holbert, M.A.; Tarrant, M.K.; Curtet, S.; Colquhoun, D.R.; Dancy, B.M.; Dancy, B.C.; Hwang, Y.; Tang, Y.; Meeth, K.; et al. Site-Specific Introduction of an Acetyl-Lysine Mimic into Peptides and Proteins by Cysteine Alkylation. J. Am. Chem. Soc. 2010, 132, 9986–9987. [Google Scholar] [CrossRef] [Green Version]
- Le, D.D.; Cortesi, A.T.; Myers, S.A.; Burlingame, A.L.; Fujimori, D.G. Site-Specific and Regiospecific Installation of Methylarginine Analogues into Recombinant Histones and Insights into Effector Protein Binding. J. Am. Chem. Soc. 2013, 135, 2879–2882. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 6, 425–479. [Google Scholar] [CrossRef]
- Weller, C.E.; Pilkerton, M.E.; Chatterjee, C. Chemical Strategies to Understand the Language of Ubiquitin Signaling. Biopolymers 2014, 101, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jbara, M.; Sun, H.; Kamnesky, G.; Brik, A. Chemical Chromatin Ubiquitylation. Curr. Opin. Chem. Biol. 2018, 45, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ai, Y.; Wang, J.; Haracska, L.; Zhuang, Z. Chemically Ubiquitylated PCNA as a Probe for Eukaryotic Translesion DNA Synthesis. Nat. Chem. Biol. 2010, 6, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; McGinty, R.K.; Fierz, B.; Muir, T.W. Disulfide-Directed Histone Ubiquitylation Reveals Plasticity in HDot1L Activation. Nat. Chem. Biol. 2010, 6, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Valkevich, E.M.; Guenette, R.G.; Sanchez, N.A.; Chen, Y.C.; Ge, Y.; Strieter, E.R. Forging Isopeptide Bonds Using Thiol-Ene Chemistry: Site-Specific Coupling of Ubiquitin Molecules for Studying the Activity of Isopeptidases. J. Am. Chem. Soc. 2012, 134, 6916–6919. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Krantz, B.; Russell, N.S.; Deshpande, S.; Wilkinson, K.D. Nonhydrolyzable Diubiquitin Analogues Are Inhibitors of Ubiquitin Conjugation and Deconjugation. Biochemistry 2000, 39, 10001–10010. [Google Scholar] [CrossRef]
- Morgan, M.; Jbara, M.; Brik, A.; Wolberger, C. Semisynthesis of Ubiquitinated Histone H2B with a Native or Nonhydrolyzable Linkage. Methods Enzymol. 2019, 618, 1–27. [Google Scholar]
- Long, L.; Furgason, M.; Yao, T. Generation of Nonhydrolyzable Ubiquitin-Histone Mimics. Methods 2014, 70, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Sahu, I.; Mali, S.M.; Hemantha, H.P.; Kleifeld, O.; Glickman, M.H.; Brik, A. Synthetic Uncleavable Ubiquitinated Proteins Dissect Proteasome Deubiquitination and Degradation, and Highlight Distinctive Fate of Tetraubiquitin. J. Am. Chem. Soc. 2016, 138, 16004–16015. [Google Scholar] [CrossRef]
- Chu, G.C.; Pan, M.; Li, J.; Liu, S.; Zuo, C.; Tong, Z.B.; Bai, J.S.; Gong, Q.; Ai, H.; Fan, J.; et al. Cysteine-Aminoethylation-Assisted Chemical Ubiquitination of Recombinant Histones. J. Am. Chem. Soc. 2019, 141, 3654–3663. [Google Scholar] [CrossRef]
- Chalker, J.M.; Gunnoo, S.B.; Boutureira, O.; Gerstberger, S.C.; Fernández-González, M.; Bernardes, G.J.L.; Griffin, L.; Hailu, H.; Schofield, C.J.; Davis, B.G. Methods for Converting Cysteine to Dehydroalanine on Peptides and Proteins. Chem. Sci. 2011, 2, 1666–1676. [Google Scholar] [CrossRef]
- Guo, J.; Wang, J.; Lee, J.S.; Schultz, P.G. Site-Specific Incorporation of Methyl- and Acetyl-Lysine Analogues into Recombinant Proteins. Angew. Chem. Int. Ed. 2008, 47, 6399–6401. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.M.; Lercher, L.; Rose, N.R.; Schofield, C.J.; Davis, B.G. Conversion of Cysteine into Dehydroalanine Enables Access to Synthetic Histones Bearing Diverse Post-Translational Modifications. Angew. Chem. Int. Ed. 2012, 51, 1835–1839. [Google Scholar] [CrossRef]
- Lercher, L.; Raj, R.; Patel, N.A.; Price, J.; Mohammed, S.; Robinson, C.V.; Schofield, C.J.; Davis, B.G. Generation of a Synthetic GlcNAcylated Nucleosome Reveals Regulation of Stability by H2A-Thr101 GlcNAcylation. Nat. Commun. 2015, 6, 7978. [Google Scholar] [CrossRef] [Green Version]
- Raj, R.; Lercher, L.; Mohammed, S.; Davis, B.G. Synthetic Nucleosomes Reveal That GlcNAcylation Modulates Direct Interaction with the FACT Complex. Angew. Chem. Int. Ed. 2016, 55, 8918–8922. [Google Scholar] [CrossRef] [Green Version]
- Lindstedt, P.R.; Taylor, R.J.; Bernardes, G.J.L.; Vendruscolo, M. Facile Installation of Post-Translational Modifications on the Tau Protein via Chemical Mutagenesis. ACS Chem. Neurosci. 2021, 12, 557–561. [Google Scholar] [CrossRef]
- Chooi, K.P.; Galan, S.R.G.; Raj, R.; McCullagh, J.; Mohammed, S.; Jones, L.H.; Davis, B.G. Synthetic Phosphorylation of P38α Recapitulates Protein Kinase Activity. J. Am. Chem. Soc. 2014, 136, 1698–1701. [Google Scholar] [CrossRef]
- Meledin, R.; Mali, S.M.; Singh, S.K.; Brik, A. Protein Ubiquitination: Via Dehydroalanine: Development and Insights into the Diastereoselective 1,4-Addition Step. Org. Biomol. Chem. 2016, 14, 4817–4823. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.H.; Bower, B.J.; Chalker, J.M.; Bernardes, G.J.L.; Wiewiora, R.; Ng, W.L.; Raj, R.; Faulkner, S.; Vallée, M.R.J.; Phanumartwiwath, A.; et al. Posttranslational Mutagenesis: A Chemical Strategy for Exploring Protein Side-Chain Diversity. Science 2016, 354, aag1465. [Google Scholar] [CrossRef]
- Yang, A.; Ha, S.; Ahn, J.; Kim, R.; Kim, S.; Lee, Y.; Kim, J.; Söll, D.; Lee, H.Y.; Park, H.S. A Chemical Biology Route to Site-Specific Authentic Protein Modifications. Science 2016, 354, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Josephson, B.; Fehl, C.; Isenegger, P.G.; Nadal, S.; Wright, T.H.; Poh, A.W.J.; Bower, B.J.; Giltrap, A.M.; Chen, L.; Batchelor-McAuley, C.; et al. Light-Driven Post-Translational Installation of Reactive Protein Side Chains. Nature 2020, 585, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R.C.; Smith, F.R.; Long, J.E.; Scott, D.; Williams, H.E.L.; Oldham, N.J.; Layfield, R.; Mitchell, N.J. Site-Selective Installation of N ϵ -Modified Sidechains into Peptide and Protein Scaffolds via Visible-Light-Mediated Desulfurative C–C Bond Formation. Angew. Chem. Int. Ed. 2022, 134, e202110223. [Google Scholar] [CrossRef]
- Willwacher, J.; Raj, R.; Mohammed, S.; Davis, B.G. Selective Metal-Site-Guided Arylation of Proteins. J. Am. Chem. Soc. 2016, 138, 8678–8681. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, J.B.; Terenzi, H.; Hüttelmaier, S.; Bernardes, G.J.L. Posttranslational Chemical Mutagenesis: To Reveal the Role of Noncatalytic Cysteine Residues in Pathogenic Bacterial Phosphatases. Biochemistry 2018, 57, 6144–6152. [Google Scholar] [CrossRef]
- Whedon, S.D.; Markandeya, N.; Rana, A.S.J.B.; Senger, N.A.; Weller, C.E.; Tureček, F.; Strieter, E.R.; Chatterjee, C. Selenocysteine as a Latent Bioorthogonal Electrophilic Probe for Deubiquitylating Enzymes. J. Am. Chem. Soc. 2016, 138, 13774–13777. [Google Scholar] [CrossRef]
- Sun, H.; Mali, S.M.; Singh, S.K.; Meledin, R.; Brik, A.; Kwon, Y.T.; Kravtsova-Ivantsiv, Y.; Bercovich, B.; Ciechanover, A. Diverse Fate of Ubiquitin Chain Moieties: The Proximal Is Degraded with the Target, and the Distal Protects the Proximal from Removal and Recycles. Proc. Natl. Acad. Sci. USA 2019, 116, 7805–7812. [Google Scholar] [CrossRef] [Green Version]
- Pomplun, S.; Jbara, M.; Schissel, C.K.; Wilson Hawken, S.; Boija, A.; Li, C.; Klein, I.; Pentelute, B.L. Parallel Automated Flow Synthesis of Covalent Protein Complexes That Can Inhibit MYC-Driven Transcription. ACS Cent. Sci. 2021, 7, 1408–1418. [Google Scholar] [CrossRef]
- Fan, C.; Deng, Q.; Zhu, T.F. Bioorthogonal Information Storage in L-DNA with a High-Fidelity Mirror-Image Pfu DNA Polymerase. Nat. Biotechnol. 2021, 39, 1548–1555. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harel, O.; Jbara, M. Posttranslational Chemical Mutagenesis Methods to Insert Posttranslational Modifications into Recombinant Proteins. Molecules 2022, 27, 4389. https://doi.org/10.3390/molecules27144389
Harel O, Jbara M. Posttranslational Chemical Mutagenesis Methods to Insert Posttranslational Modifications into Recombinant Proteins. Molecules. 2022; 27(14):4389. https://doi.org/10.3390/molecules27144389
Chicago/Turabian StyleHarel, Omer, and Muhammad Jbara. 2022. "Posttranslational Chemical Mutagenesis Methods to Insert Posttranslational Modifications into Recombinant Proteins" Molecules 27, no. 14: 4389. https://doi.org/10.3390/molecules27144389