
Molecular Simulation to Explore the Dissolution Behavior of Sulfur in Carbon Disulfide

Abstract
:1. Introduction
2. Model Construction and Simulation Method
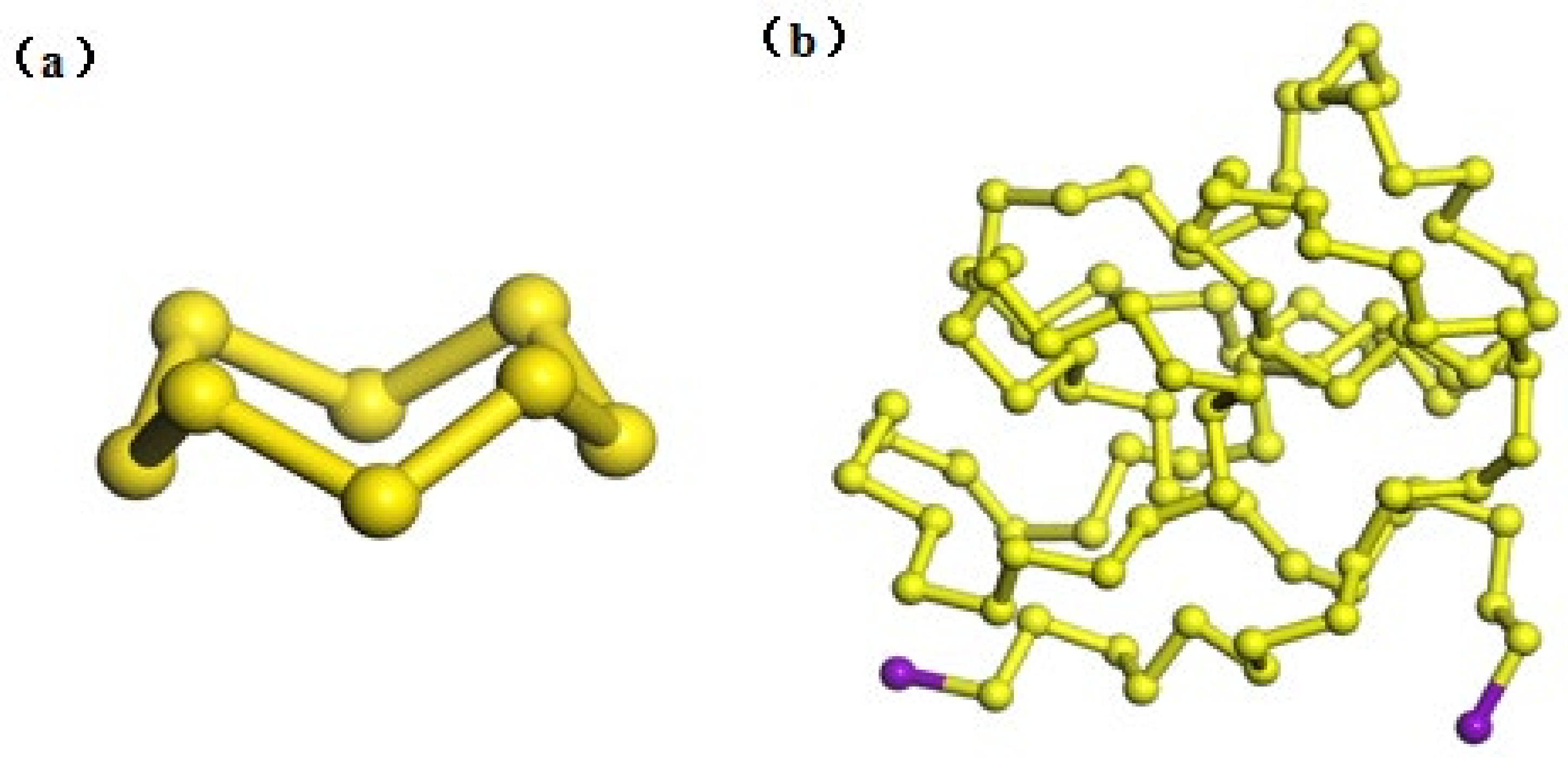
2.1. Model Construction
2.2. Simulation Details
3. Results and Discussion
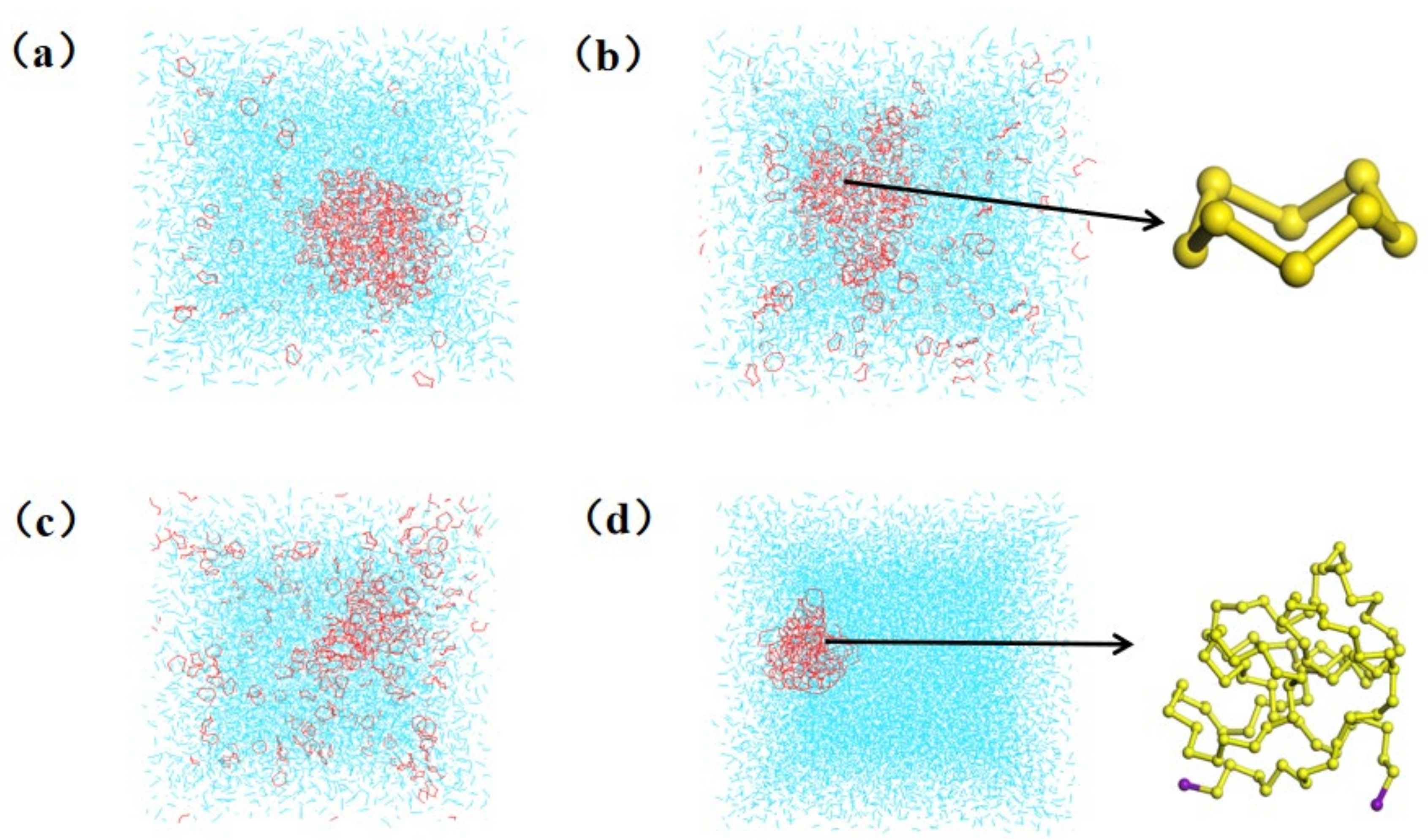
3.1. Simulation Results
3.2. Solubility Analysis of Soluble Sulfur
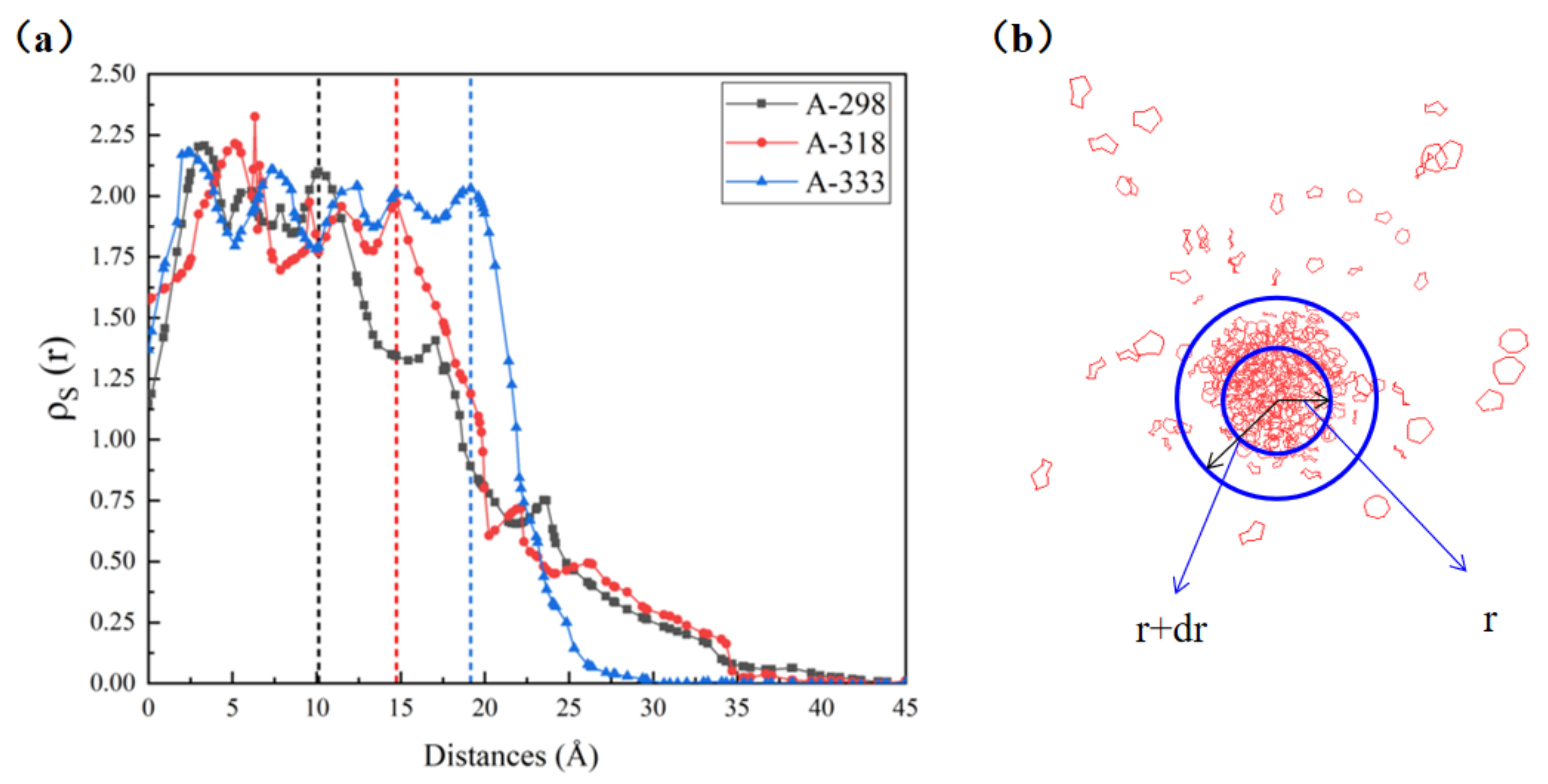
3.2.1. Properties of Dissolved Sulfur
3.2.2. Dissolved Sulfur Interaction
3.3. Solubility Analysis of Insoluble Sulfur
3.3.1. Dissolution Behavior of Insoluble Sulfur
3.3.2. Solute Interaction with Solvent
3.4. Solubility Parameter Theory
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Griebel, J.; Glass, R.S.; Char, K.; Pyun, J. Polymerizations with elemental sulfur: A novel route to high sulfur content polymers for sustainability, energy and defense. Prog. Polym. Sci. 2016, 58, 90–125. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, R.G.; Paria, S. Synthesis of sulfur nanoparticles in aqueous surfactant solutions. J. Colloid Interface Sci. 2010, 343, 439–446. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Qiu, L. Application of domestic insoluble sulfur OT-20in TBR tire. Tire Ind. 2009, 29, 550–553. [Google Scholar]
- Yuan, Y.; Zhang, X.; Song, T.; Gao, X. Application of Insoluble Sulfur Crystex Cure Pro in All-steel Radial Tire. Rubber Sci. Technol. 2022, 20, 75–78. [Google Scholar]
- Luo, H.K.; Li, D.G.; Luo, S. The effect of halide and the coordination geometry of chromium center in homogeneous catalyst system for ethylene trimerization. J. Mol. Catal. A Chem. 2004, 1–2, 75–78. [Google Scholar]
- Zhang, K.; Xia, X.; Qiang, Y. Effect of Styrene on The Extraction of Insoluble Sulfur. Spec. Petrochem. 2012, 29, 76–79. [Google Scholar]
- Jones, R.O.; Ballone, P. Density functional and Monte Carlo studies of sulfur(I): Structure and bonding in Sn rings and chains (n = 2−18). J. Phys. Chem. 2003, 118, 9257–9265. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.O.; Ballone, P. Density functional and Monte Carlo studies of sulfur(II): Equilibrium polymerization of the liquid phase. J. Phys. Chem. 2003, 119, 8704–8715. [Google Scholar]
- Wang, R.; Shen, B.; Ma, J.; Zhao, R. Ring-open reaction mechanism of sulfur S8 based on density functional theory. CIESC J. 2015, 66, 3919–3924. [Google Scholar]
- Jian, M.; Jigang, Z.; Rongjie, W.; Yang, S.; Benxian, S. Molecular simulation studies on the properties of insoluble sulfur stabilizers. Chem. Ind. Eng. Prog. 2016, 35, 706–710. [Google Scholar]
- Jigang, Z.; Yang, S.; Rongjie, W.; Benxian, S. A novel and easy way to improve the thermal stability of insoluble sulfur by curing process. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 431–436. [Google Scholar] [CrossRef]
- Audran, G.; Brémond , P.; Marque, S.R.; Santelli, M. Theoretical investigations on the conversions of cyclic polysulfides to acyclic polysulfide diradicals and subsequent reactions of biological interest. Tetrahedron 2017, 73, 3492–3496. [Google Scholar] [CrossRef]
- Orthous-Daunay, F.R.; Quirico, E.; Lemelle, L.; Beck, P.; de Andrade, V.; Simionovici, A.; Derenne, S. Speciation of sulfur in the insoluble organic matter from carbonaceous chondrites by XANES spectroscopy. Earth Planet. Sci. Lett. 2010, 300, 321–328. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Henon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Foster, R.B. Sulfur-Challenging Market Conditions Reshaping the Future. EMJ- Eng. Min. J. 1993, 194, 52–56. [Google Scholar]
- Chen, H.; Liu, C.; Xu, X. Molecular dynamic simulation of sulfur solubility in H2S system. Int. J. Mod. Phys. B. 2019, 33, 0052. [Google Scholar] [CrossRef]
- Baidakov, V.G.; Chernykh, G.G.; Protsenko, S.P. Liquid-vapor equilibrium and surface tension in Lennard-Jones systems. Russ. J. Phys. Chem. A 2000, 74, 1241–1245. [Google Scholar]
- Zhang, S.; Li, C.; Wu, Q.; Xie, Y.; Zhang, S.; Cao, Z.; Ding, S.; Yu, Q.; Wang, M.; Wang, G.; et al. Chemical Handbook; Shandong Science and Technology Press: Shandong, China, 1986; pp. 19–84. [Google Scholar]
- Lu, T.; Chen, F.; Multiwfn. A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, H.; Wang, X.; Zhang, H.; Zhang, Z.; Yuan, S. Molecular insights into the uptake of SiO2 nanoparticles on phospholipid membrane: Effect of surface properties and particle size. Colloids Surf. B. Biointerfaces 2022, 210, 112250. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, Z.; Zhang, W.; Gao, Q.; Tang, Z.; Zhao, W. Understanding the top-down fragmentation of 2D material in miscible liquid environment based on Hansen solubility parameters theory. FlatChem 2022, 32, 100346. [Google Scholar] [CrossRef]
- Han, K.H.; Jeon, G.S.; Hong, I.K.; Lee, S.B. Prediction of solubility parameter from intrinsic viscosity. J. Ind. Eng. Chem. 2013, 19, 1130–1136. [Google Scholar] [CrossRef]
- Li, G.; Li, J.; Li, H.; Gao, M.; Liang, S. Molecular Dynamics Calculation of the Solubility Parameters of Ethyl Cellulose. Guangdong Chem. Ind. 2015, 42, 5–10. [Google Scholar]
- Hildebrand, J.H.; Scott, R.L. The solubility of nonelectrolytes. Journal of chemical education; Dover Publications: Mineola, NY, USA, 1965; p. 4. [Google Scholar]
- Gao, B. Application for Solubility Parameter. North China Insitute Technol. 1998, 2, 18–20. [Google Scholar]
- Xia, Q.; Yin, K. Calculation of Solubility Parameters of Organic Solvents by Molecular Dynamics Simulation. J. Jiangsu Inst. Technol. 2004, 1, 40–42. [Google Scholar]
- Liu, G.; Hu, Y. Molecular Structure and Solubility Parameter Liquids. East China Inst. Chem. Technol. 1990, 3, 257–264. [Google Scholar]
- Li, Y.; Guo, P. Calculation of solubility parameter for liquids. J. Xinyang Norm. Univ. 2002, 1, 52–53. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance Name | Simulation Value/(MPa1/2) | Reference Value/(MPa1/2) | Relative Error/% |
|---|---|---|---|
| C6H6 | 17.6 | 19.5 | 9.7 |
| CH3COOC2H5 | 17.4 | 18.8 | 7.4 |
| CS2 | 20.6 | 19.4 | 5.8 |
| Substance Name | Simulation Value/(MPa1/2) |
|---|---|
| S8 | 29.943 |
| IS | 38.631 |
| CS2 | 21.678 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, X.; Wang, W.; Du, M.; Ma, D.; Zhang, X. Molecular Simulation to Explore the Dissolution Behavior of Sulfur in Carbon Disulfide. Molecules 2022, 27, 4402. https://doi.org/10.3390/molecules27144402
Cui X, Wang W, Du M, Ma D, Zhang X. Molecular Simulation to Explore the Dissolution Behavior of Sulfur in Carbon Disulfide. Molecules. 2022; 27(14):4402. https://doi.org/10.3390/molecules27144402
Chicago/Turabian StyleCui, Xiangyu, Wenbo Wang, Mengcheng Du, Delong Ma, and Xiaolai Zhang. 2022. "Molecular Simulation to Explore the Dissolution Behavior of Sulfur in Carbon Disulfide" Molecules 27, no. 14: 4402. https://doi.org/10.3390/molecules27144402