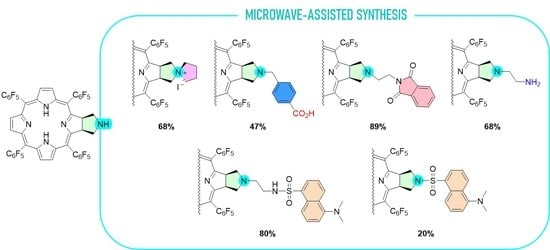
3.2. Synthesis
5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (
TPFPP) [
27] and
N-(2-aminoethyl)-5-(dimethylamino)naphthalene-1-sulfonamide (
dansylNH(CH2)2NH2) [
28] were prepared using previously reported methods.
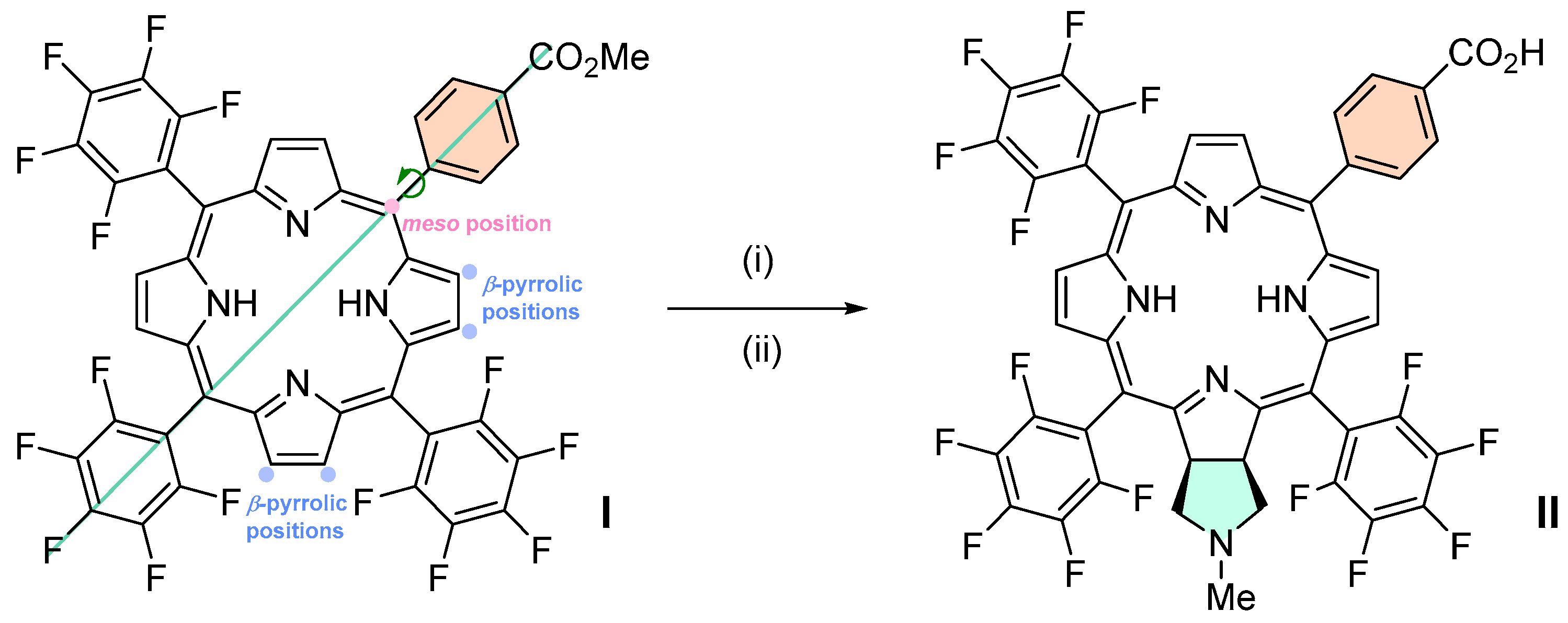
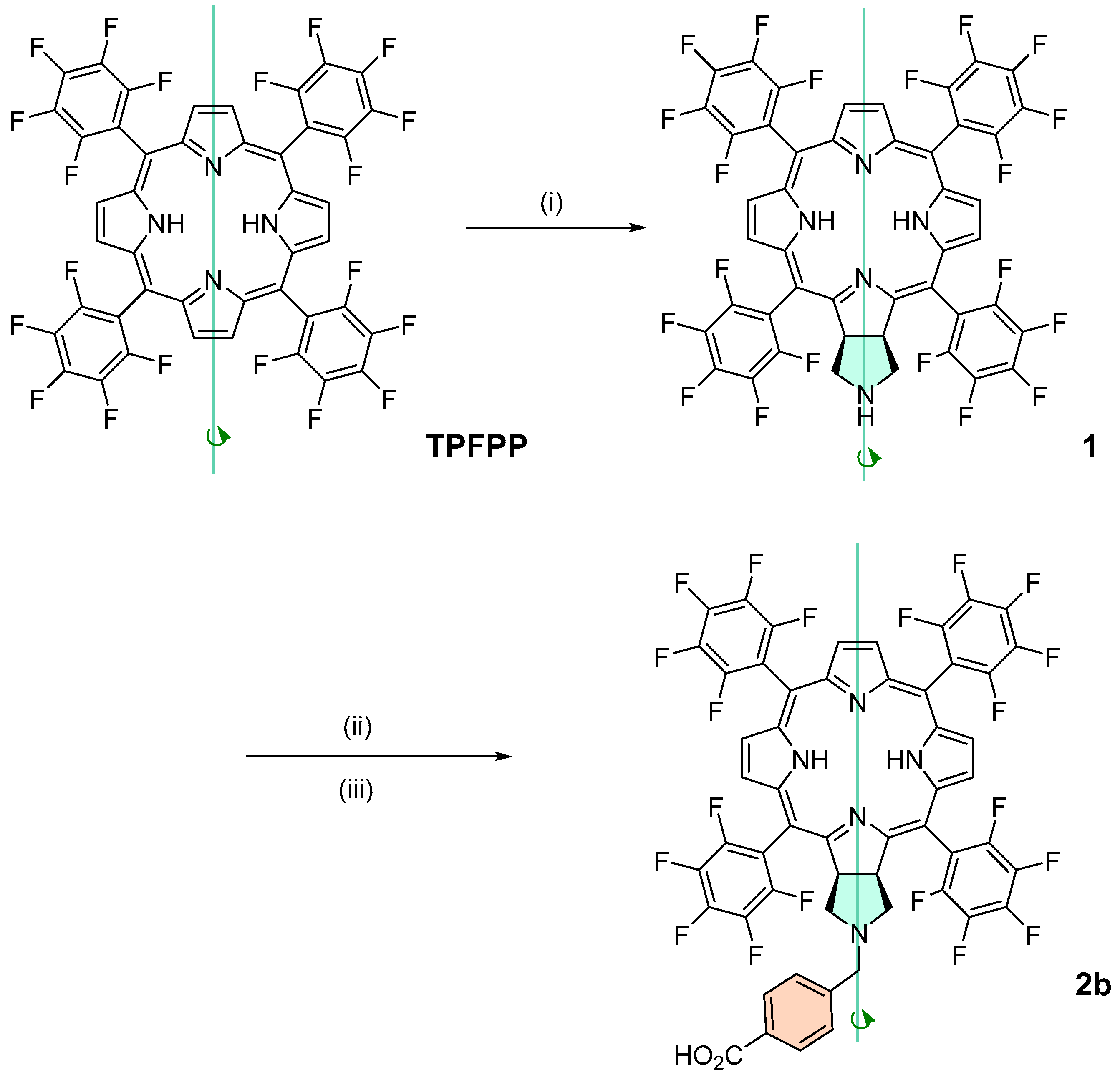
Chlorin 1 was synthesized according to a published protocol [
14] with slight modifications using two different heating methods.
3.2.1. Synthesis of Chlorin 1 under Conventional Heating
TPFPP (195 mg, 200 μmol) was dissolved in chlorobenzene (16 mL) and the solution was purged with N
2 and then stirred at 140 °C under N
2 atmosphere. After 5 min at 140 °C, 25 mg of a ground mixture of glycine (77 mg, 1.0 mmol) and paraformaldehyde (31 mg, 1.0 mmol) was transferred to the porphyrin solution and the mixture left reacting for 2 h at 140 °C under N
2 atmosphere. Three more additions of the glycine/paraformaldehyde mixture were performed every 2 h to complete a total of 8 h of reaction. After reaction completion, chlorobenzene was evaporated and the reaction mixture was dissolved in CH
2Cl
2 and purified by flash chromatography using CH
2Cl
2/acetone (98:2) as the eluent to isolate the unchanged
TPFPP (38% recovery) and minor products; then CH
2Cl
2/acetone (90:10) was used to isolate the major green fraction. After solvent evaporation, the residue corresponding to the major fraction was stirred with a mixture of TFA/H
2O (9:1) (50 mL) at room temperature for 3 h. The mixture was neutralized with a saturated solution of NaHCO
3 and washed with deionized water. The organic extract was dried (Na
2SO
4) and filtered and the solvent evaporated to obtain a yield of 50% chlorin
1. Spectroscopic data are as reported in [
14].
1H NMR (400.14 MHz, CDCl
3) δ −1.83 (s, 2H, NH), 3.16 (dd,
J = 11.5, and 3.9 Hz, 2H, CH
2-pyrrolidine), 3.30–3.50 (m, 2H, CH
2-pyrrolidine), 5.21–5.24 (m, 2H, H-2,3), 8.40 (d,
J = 5.0 Hz, 2H), 8.49 (s, 2H, H-12,13), 8.72 (d,
J = 5.0 Hz, 2H).
3.2.2. Synthesis of Chlorin 1 under Microwave Heating
TPFPP (200 mg, 205 μmol) was transferred to a microwavable 30 mL glass vessel and dissolved in chlorobenzene (16 mL). The solution was then purged with N2. A mixture of glycine (77 mg, 1.0 mmol) and paraformaldehyde (31 mg, 1.0 mmol) was ground and 25 mg of it was added to the TPFPP solution. The vessel was placed inside the microwave reactor and it was irradiated at a maximum microwave power of 290 W until reaching 135 °C; the power was modulated for 1 h at constant temperature. Three more additions of the ground mixture were performed to complete a total of 4 h of reaction. After reaction completion, the chlorobenzene was evaporated, and the reaction mixture was dissolved in CH2Cl2 and purified by flash chromatography using CH2Cl2 to isolate the unchanged TPFPP (30% recovered) and other minor products and then CH2Cl2/methanol (99:1) to isolate the major green fraction. After solvent evaporation, the residue corresponding to the major fraction was stirred with a mixture of TFA/H2O (9:1) (50 mL) at room temperature for 3 h. Then it was neutralized with a saturated solution of NaHCO3 and washed with deionized water. The organic extract was dried (Na2SO4), filtered and the solvent evaporated to obtain a 41% yield of chlorin 1.
3.2.3. Synthesis of Chlorin Zn-1
Chlorin 1 (20 mg, 20 μmol), CH3CN (2 mL), and Zn(II) acetate dihydrate (43.8 mg, 200 μmol) were transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. After bubbling the solution with N2, the vessel was sealed with a silicone septum and placed into the cavity of the microwave reactor. The reaction mixture was then heated to 120 °C using a maximum power of 100 W, which was automatically modulated for 1 min. After this time, the solution was washed several times with water. The organic layer was dried (Na2SO4) and the solvent evaporated. Chlorin Zn-1 was obtained in quantitative yield. 1H NMR (400.14 MHz, DMSO-d6) δ 8.78 (d, J = 4.8 Hz, 2H), 8.59 (s, 2H), 8.45–8.39 (m, 2H), 5.01 (m, 2H), 2.55 (m, 2H, overlapped with solvent signal), and 2.45 (m, 2H, overlapped with solvent signal). UV-Vis (DMF) λmax (ε) 416 (34 × 103); 517 (very low intensity); 578 (very low intensity); 620 (5.6 × 103) nm. Fluorescence (DMF) λmax 625; 680 nm.
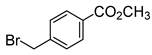
3.2.4. Synthesis of Chlorin 2a and 2b
A solution of chlorin 1 (50.3 mg, 49.4 μmol) in anhydrous DMF (2 mL) was transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar and methyl 4-(bromomethyl)benzoate (33.9 mg, 148 μmol) and DIPEA (27.5 μL, 287 nmol) were added. The vessel was sealed with a silicone septum and placed into the microwave cavity. The reaction mixture was then heated to 75 °C using a maximum microwave power of 50 W, which was automatically modulated for 5 min. The solution was afterwards poured into 40 mL of deionized water, and the resulting chlorin 2a was obtained as a green precipitate, which was filtered and recrystallized in CHCl3/MeOH, and we proceeded directly to the ester hydrolysis. Then, the resulting chlorin 2a was dissolved in TFA (5 mL) and HCl (10 mL) and the resulting mixture was heated to 85 °C for 24 h. After this time, it was neutralized with a saturated solution of Na2CO3, extracted with CH2Cl2, and washed with deionized water. The organic extract was dried (Na2SO4) and filtered and the solvent evaporated to yield 27 mg of chlorin 2b (a 47% yield). 1H NMR (400.14 MHz, CD3OD) δ 2.71–2.89 (m, 2H, CH2-pyrrolidine), 3.05–3.16 (m, 2H, CH2-pyrrolidine), 3.53 (s, 2H, -CH2-ArCOOH), 5.25–5.37 (m, 2H, H-2,3), 7.15 (d, J = 8.2 Hz, 2H, Ar-COOH), 7.86 (d, J = 8.2 Hz, 2H, Ar-COOH), 8.70–8.59 (m, 2H, H-β), 8.99 (d, J = 5.1 Hz, 2H, H-β). 19F{1H} NMR (376.46 MHz, CD3OD) δ (ppm): −165.13 (dddd, J = 28.0, 20.0, 23.0 and 7.9 Hz, 4F, Fmeta-Ar), −164.06 (ddd, J = 23.4, 20.6 and 8.0 Hz, 2F, Fmeta-Ar), −163.52 (ddd, J = 23.4, 20.1 and 8.2 Hz, 2F, Fmeta-Ar), −155.80 (t, J = 20.0 Hz, 2F, Fpara-Ar), −155.33 (t, J = 20.0 Hz, 2F, Fpara-Ar), −140.67 to −140.41 (m, 6F, Fortho-Ar), −137.98 (dd, J = 23.7 and 7.6 Hz, 2F, Fortho-Ar). UV-Vis (DMF) λmax (ε) 405 (120 × 103); 503 (22 × 103); 598 (5.8 × 103); 650 (60 × 103) nm. Fluorescence (DMF) λmax 655; 717 nm; ϕF = 0.101. HRMS (ESI) m/z: [M+H]+ Calcd for C54H22F20N5O2+ 1152.1449, found 1152.1507.
3.2.5. Synthesis of Chlorin 2c
Chlorin 2b (15.9 mg, 13.8 μmol) and anhydrous DMF (100 μL) were transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. To this solution were then added 1-hydroxybenzotriazole monohydrate (HOBt, 1.8 mg, 13 μmol), N,N-diisopropylethylamine (DIPEA, 5.0 μL, 13 μmol), 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDC, 2.5 mg, 13 μmol), and aniline (3.0 μL, 33 μmol). The vessel was sealed with a silicone septum in a N2 atmosphere and placed in the cavity of the microwave reactor. The reaction mixture was then heated to 75 °C using a maximum microwave power of 100 W, which was automatically modulated for 20 min. The reaction mixture was washed with water once and the organic layer was extracted with ethyl acetate. The organic solvent was partially evaporated and the residue was chromatographed in a silica gel column using a mixture of toluene:ethyl acetate (7:3). The first eluted fraction was the activated ester, then chlorin 2c (3.4 mg, 20% yield), then chlorin 2b. 1H NMR (400.14 MHz, CDCl3) δ (ppm): −1.80 (s, 2H, NH), 2.66–2.73 (m, 2H, CH2-pyrrolidine), 3.03–3.12 (m, 2H, CH2-pyrrolidine), 3.51 (s, 2H, -CH2-ArCONH-Ar), 5.19–5.23 (m, 2H, H-2,3), 7.16 (d, J = 7.9 Hz, 2H, Ar-CONH-Ar), 7.37 (m, 2H, H-aniline), 7.59–7.66 (m, 2H, aniline), 7.67–7.75 (m, 2H, Ar-CONH-Ar + 2H, Hpara-aniline), 8.38 (d, J = 5.0 Hz, 2H, H-β), 8.49 (s, 2H, H-β), 8.72 (d, J = 5.0 Hz, 2H, H-β). 13C NMR (100.63 MHz, CDCl3) δ (ppm): 29.9, 52.5, 60.7, 120.3, 124.0, 127.2, 128.2, 129.3, 132.5, 134.3, 135.4, 140.5, 152.9, 165.4, 169.0. HRMS (ESI) m/z: [M+H]+ Calcd for C60H27F20N6O+ 1227.1921, found 1227.2080.
3.2.6. Synthesis of Chlorin 3
Chlorin 1 (50 mg, 49 μmol), DMF (3 mL) and DIPEA (9.4 μL, 54 μmol) were transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. Immediately, 1,4-diiodobutane (7.1 μL, 54 μmol) was added to the resulting solution and the vessel was sealed with a silicone septum and placed into the microwave cavity. The reaction mixture was then heated to 75 °C using a maximum microwave power of 50 W, which was automatically modulated for 30 min. After this time, one more addition of 1,4-diiodobutane (7.1 μL, 54 μmol) was performed and the reaction followed the same procedure as described previously. The reaction mixture was then diluted with ethyl acetate and washed four times with deionized water, and the organic extract was dried (Na2SO4), filtered, and concentrated. The resulting residue was dissolved in CH2Cl2 and chromatographed (silica column) using a mixture of CH2Cl2/MeOH (98:2) to elute the starting chlorin 1 (~10%). The eluent was changed to CH2Cl2/MeOH (95:5) to elute chlorin 3. After crystallization from CH2Cl2/hexane, 40 mg were obtained (68% yield). 1H NMR (400.14 MHz, CD3OD) δ 2.11 (quint, J = 7.4 Hz, 2H, H-3′’) 2.31 (quint, J = 7.5 Hz, 2H, H-4′’), 3.43 (t, J = 7.3 Hz, 2H, H-2′’), 3.94 (m, 4H, H-5′’ and H-21), 4.32–4.21 (m, 2H, H-31), 5.91 (t, J = 6.4 Hz, 2H, H-2,3), 8.69 (dd, J = 5.1 and 1.2 Hz, 2H, H-β), 8.71 (s, 2H, H-12,13), 9.06 (d, J = 5.1 Hz, 2H, H-β). 19F{1H} NMR (376.46 MHz, CD3OD) δ (ppm): −164.93 (dddd, J = 23.1, 19.6, 11.2 and 8.0 Hz, 4F, Fmeta-Ar), −162.48 (dddd, J = 24.1, 20.3, 15.8 and 8.1 Hz, 4F, Fmeta-Ar), −155.42 (t, J = 20.1 Hz, 2F, Fpara-Ar), −153.87 (t, J = 20.3 Hz, 2F, Fpara-Ar), −140.77 (dd, J = 22.9 and 8.8 Hz, 2F, Fortho-Ar), −140.58 (dd, J = 23.1 and 8.6 Hz, 2F, Fortho-Ar), −140.04 (dd, J = 23.4 and 8.4 Hz, 2F, Fortho-Ar), −138.91 (dd, J = 23.6 and 7.9 Hz, 2F, Fortho-Ar). 13C{1H} NMR (100.63 MHz, CD3OD) δ (ppm): 23.8, 23.9 (C-3’ and C-4’), 52.5 (C-2 and C-3-pyrrolidine), 64.4, 64.5 (C-2’ and C3’), 68.5 (C-21 and C-31), 98.1, 108.3, 115.9, 126.2, 130.3, 134.3, 137.1, 141.8, 155.0, 165.6. UV-Vis (DMF) λmax (ε) 401 (162 × 103); 500 (12 × 103); 527 (4 × 103); 595 (3 × 103); 647 (41 × 103) nm. Fluorescence (DMF) λmax 651; 716 nm; ϕF = 0.333. MS (MALDI-TOF) m/z: [M]+ Calcd for C50H22F20N5+ 1072.155, found 1072.544.
3.2.7. Synthesis of Chlorin Zn-3
Chlorin Zn-1 (16 mg, 15 μmol), DMF (1 mL) and DIPEA (50 μL, 290 μmol) were transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. Immediately, 1,4-diiodobutane (30 μL, 380 μmol) was added to the resulting solution and the vessel was sealed with a silicone septum and placed into the microwave cavity. The reaction mixture was then heated to 75 °C using a maximum microwave power of 50 W, which was automatically modulated for 30 min. The reaction mixture was then diluted with ethyl acetate and washed four times with deionized water. The organic extract was dried (Na2SO4), filtered, and concentrated. The resulting residue was dissolved in CH2Cl2 and purified by preparative thin-layer chromatography using ethyl acetate as the eluent. After crystallization from CH2Cl2/hexane, 12.6 mg of chlorin Zn-3 were obtained (68% yield). 1H NMR (400.14 MHz, DMSO-d6) δ (ppm): 1.96 (quint, J = 7.5 Hz, 2H), 2.16 (quint, J = 7.4 Hz, 2H), 3.40 (t, J = 7.5 Hz, 2H, H-52), 3.74–3.82 (m, 4H, H-22 and H-21), 4.04 (s, 1H), 5.67 (m, 2H, H-2,3), 8.42 (dd, J = 4.6 and 1.2 Hz, 2H, H-β), 8.66 (s, 2H, H-12,13), 8.84 (d, J = 4.7 Hz, 2H, H-β). 19F{1H} NMR (376.46 MHz, DMSO-d6) δ (ppm): −163.23 to −162.97 (m, 4F, Fmeta-Ar), −161.30 to −161.09 (m, 2F, Fmeta-Ar), −160.73 to −160.52 (m, 2F, Fmeta-Ar), −154.66 (t, J = 22.5 Hz, 2F, Fpara-Ar), −153.75 (t, J = 22.8 Hz, 2F, Fpara-Ar), −139.86 (td, J = 26.7 and 7.4 Hz, Fortho-Ar), −139.03 (dd, J = 26.3 and 7.5 Hz, Fortho-Ar), −137.17 (dd, J = 26.2 and 7.4 Hz, Fortho-Ar). UV-Vis (DMF) λmax (ε) 417 (200 × 103); 622 (34 × 103) nm. Fluorescence (DMF) λmax 625; 675 nm; ϕF = 0.076.
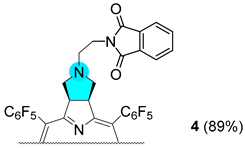
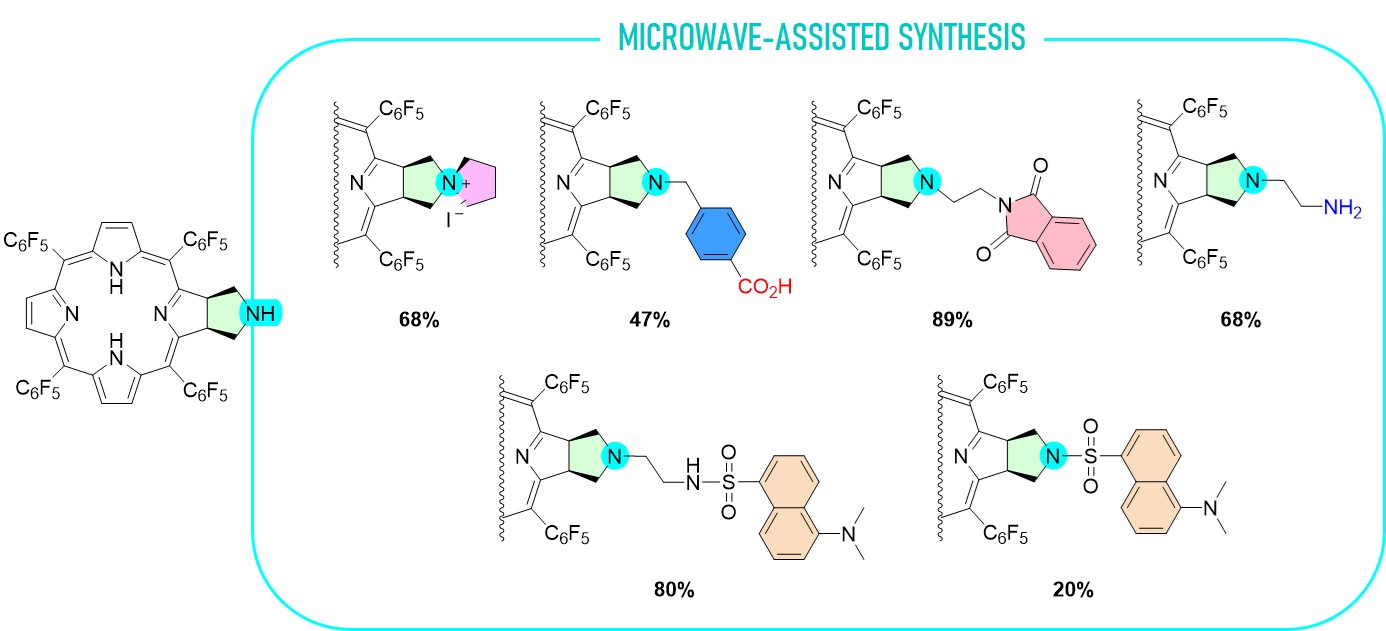
3.2.8. Synthesis of Chlorin 4
Chlorin 1 (94.2 μmol) and anhydrous DMF (150 μL) were transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. To this solution was then added N-(2-bromoethyl)phthalimide (41.9 mg, 165 μmol) and K2CO3 (29.9 mg, 217 μmol). The vessel was sealed with a silicone septum and placed into the microwave cavity. The reaction mixture was then heated to 75 °C using a maximum microwave power of 50 W, which was automatically modulated for 30 min. After this time one more addition of N-(2-bromoethyl)phthalimide (165 μmol) and K2CO3 (204 μmol) was performed. The solution was again heated to 75 °C using the same microwave reactor parameters. The reaction mixture was washed several times with water, and the organic layer was extracted with ethyl acetate. The organic phase was concentrated and chromatographed in a silica gel column using a mixture of CH2Cl2:hexane (16:4). Chlorin 4 was isolated in 89% yield (100 mg). 1H NMR (400.14 MHz, CDCl3) δ (ppm): −1.90 (s, 2H, NH) 2.54 (t, J = 6.9 Hz, 2H, CH2), 2.64 (t, J = 6.0 Hz, 2H, N-CH2CH2-Phth), 3.31 (t, J = 8.0 Hz, 2H, CH2), 3.58–3.67 (m, 2H, N-CH2CH2-Phth), 5.09 (q, J = 7.0 Hz, 2H, H-2, 3), 7.17–7.28 (m, 4H, Ar-Phth), 8.40 (d, J = 5.0 Hz, 2H, H-β), 8.50 (s, 2H, H-β), 8.71 (d, J = 5.0 Hz, 2H, H-β). 13C{1H} NMR (100.63 MHz, CDCl3) δ (ppm): 36.4 (N-CH2CH2-Phth), 51.5 (N-CH2CH2-Phth), 52.6 (CH2-pyrrolidine), 60.3 (C-N-pyrrolidine), 97.1, 106.2, 122.7 (Ar-Phth), 124.1, 128.1, 131.7 (β-pyrrole), 132.4 (Ar-Phth), 133.5, 135.4, 140.5, 152.7, 168.1, 169.2. 19F{1H} NMR (376.46 MHz, CDCl3) δ (ppm): −161.52 (dddd, J = 33.7, 24.1, 20.5 and 8.7 Hz, 4F, Fmeta-Ar), −160.58 (ddd, J = 24.1, 20.8 and 8.4 Hz, 2F, Fmeta-Ar), −160.34 (ddd, J = 24.0, 20.8 and 8.4 Hz, 2F, Fmeta-Ar), −151.77 (t, J = 20.9 Hz, 2F, Fpara-Ar), −151.38 (t, J = 20.9 Hz, 2F, Fpara-Ar), −137.82 (dd, J = 24.2 and 8.3 Hz, 2F, Fortho-Ar), −136.96 (tdd, J = 20.0, 8.8 and 3.4 Hz, 4F, Fortho-Ar), −135.44 (dd, J = 24.1 and 8.0 Hz, 2F, Fortho-Ar). HRMS (ESI) m/z: [M+H]+ Calcd for C56H23F20N6O2+ 1191.1558; Found 1191.1513.
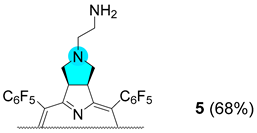
3.2.9. Synthesis of Chlorin 5
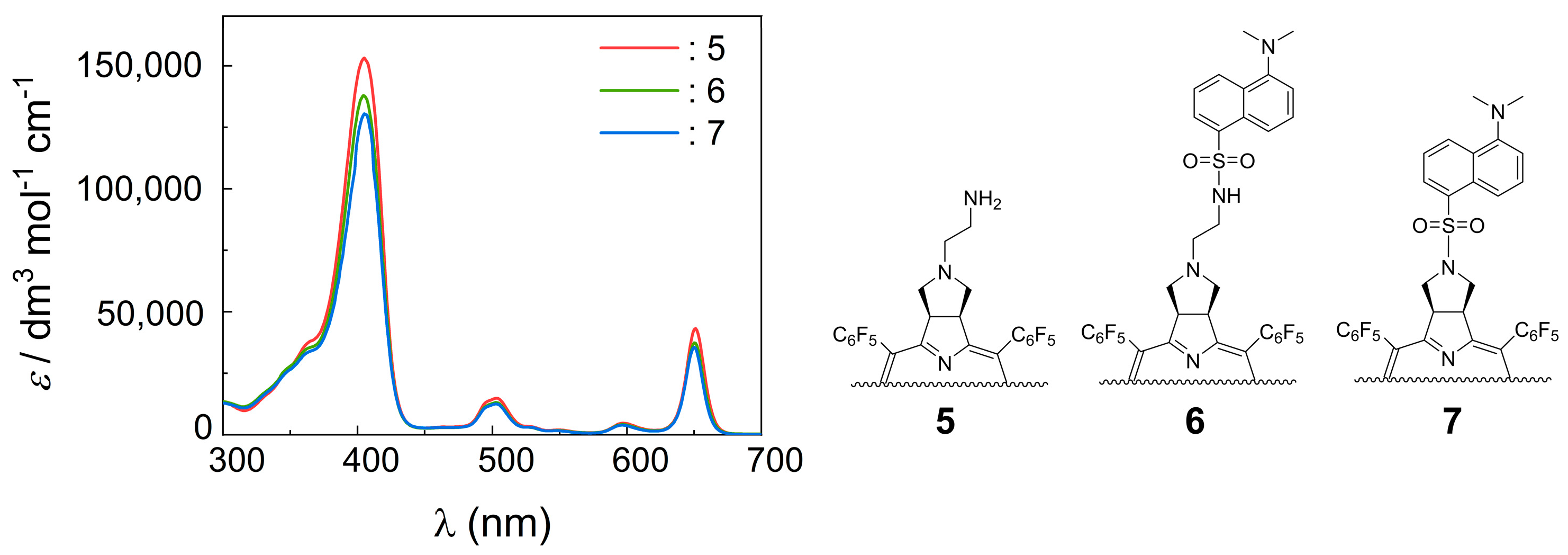
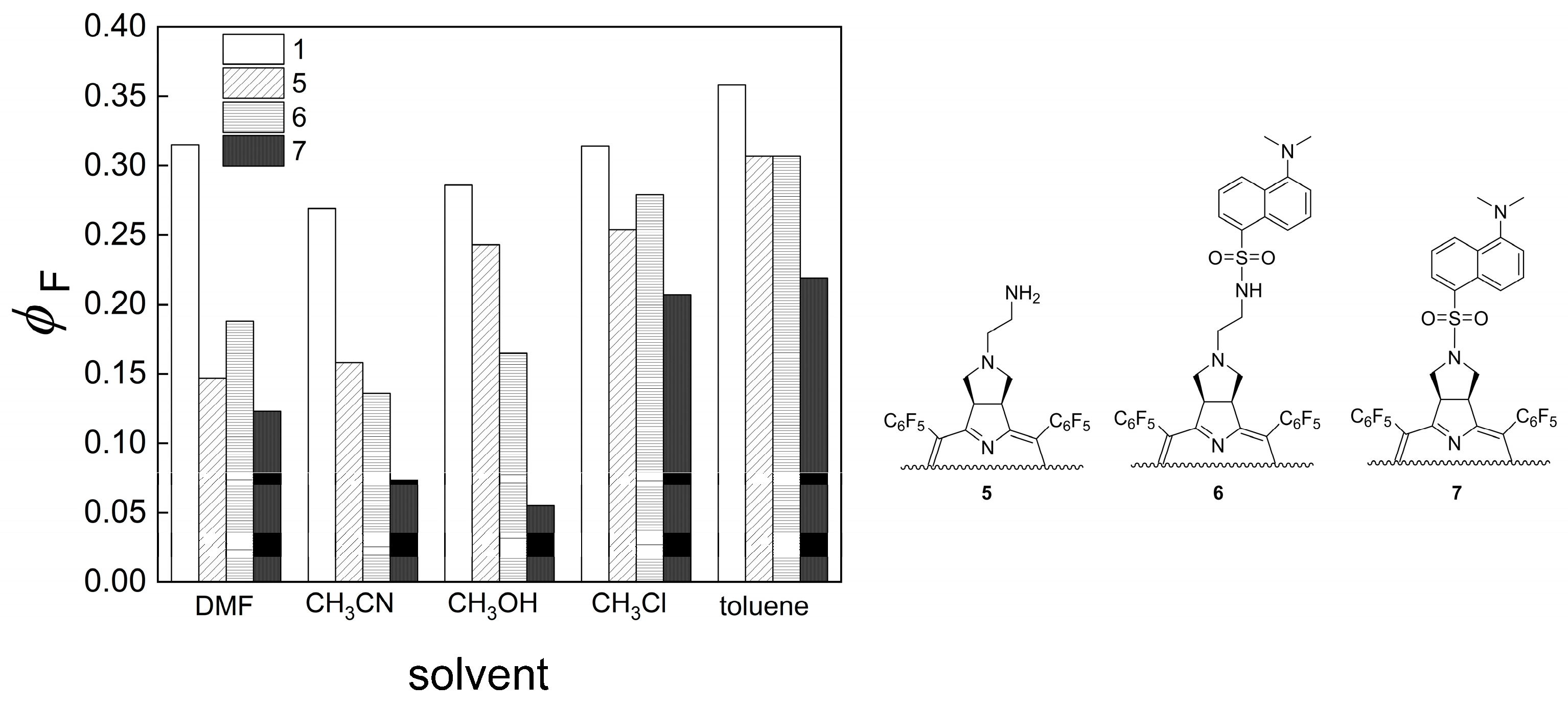
Chlorin 1 (200 mg, 197 μmol), 1 mL of DMF, K2CO3 (81.5 mg, 589 μmol) and 2-bromoethanaminium bromide (80.5 mg, 393 μmol) were placed in a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. The vessel was sealed with a silicone septum and placed into the microwave reactor cavity. The reaction mixture was then heated to 75 °C using a maximum microwave power of 50 W, which was automatically modulated for 5 min. After this time, the solution was washed with a saturated solution of NaHCO3 and deionized water. The organic extract was dried (Na2SO4), filtered, concentrated, and purified by preparative TLC using CH2Cl2/MeOH (95:5) as an eluent system, isolating 14.2 mg of chlorin 5 (68% yield). 1H NMR (400.14 MHz, CDCl3) δ (ppm): −1.83 (s, 2H, NH), 2.54 (bs, 2H), 2.65 (bs, 2H), 2.99 (bs, 2H), 5.30–5.45 (m, 2H, H-2,3), 8.39 (d, J = 5.0, 2H, H-β), 8.48 (s, 2H, H-12, 13), 8.71 (d, J = 5.0 Hz, 2H, H-β). 19F{1H} NMR (376.46 MHz, CDCl3) δ (ppm): −161.70 to −161.25 (m, 4F, Fmeta-Ar), −160.83 (td, J = 22.2 and 8.2 Hz, 2F, Fmeta-Ar), −160.25 (td, J = 22.7 and 8.2 Hz, 2F, Fmeta-Ar), −151.73 (t, J = 20.9 Hz, 2F, Fpara-Ar), −151.53 (t, J = 21.3 Hz, 2F, Fpara-Ar), −137.65 (dd, J = 24.1 and 8.2 Hz, 2F, Fortho-Ar), −136.93 (dd, J = 23.6 and 8.3 Hz, 4F, Fortho-Ar), −135.81 (dd, J = 24.4 and 8.7 Hz, 2F, Fortho-Ar). UV-Vis (DMF) λmax (ε) 405 (153 × 103); 503 (15 × 103); 597 (5 × 103); 650 (43 × 103) nm. Fluorescence (DMF) λmax 655; 717 nm; ϕF = 0.147. HRMS (ESI) m/z: [M+H]+ Calcd for C48H21F20N6+ 1061.1503; Found 1061.1513.
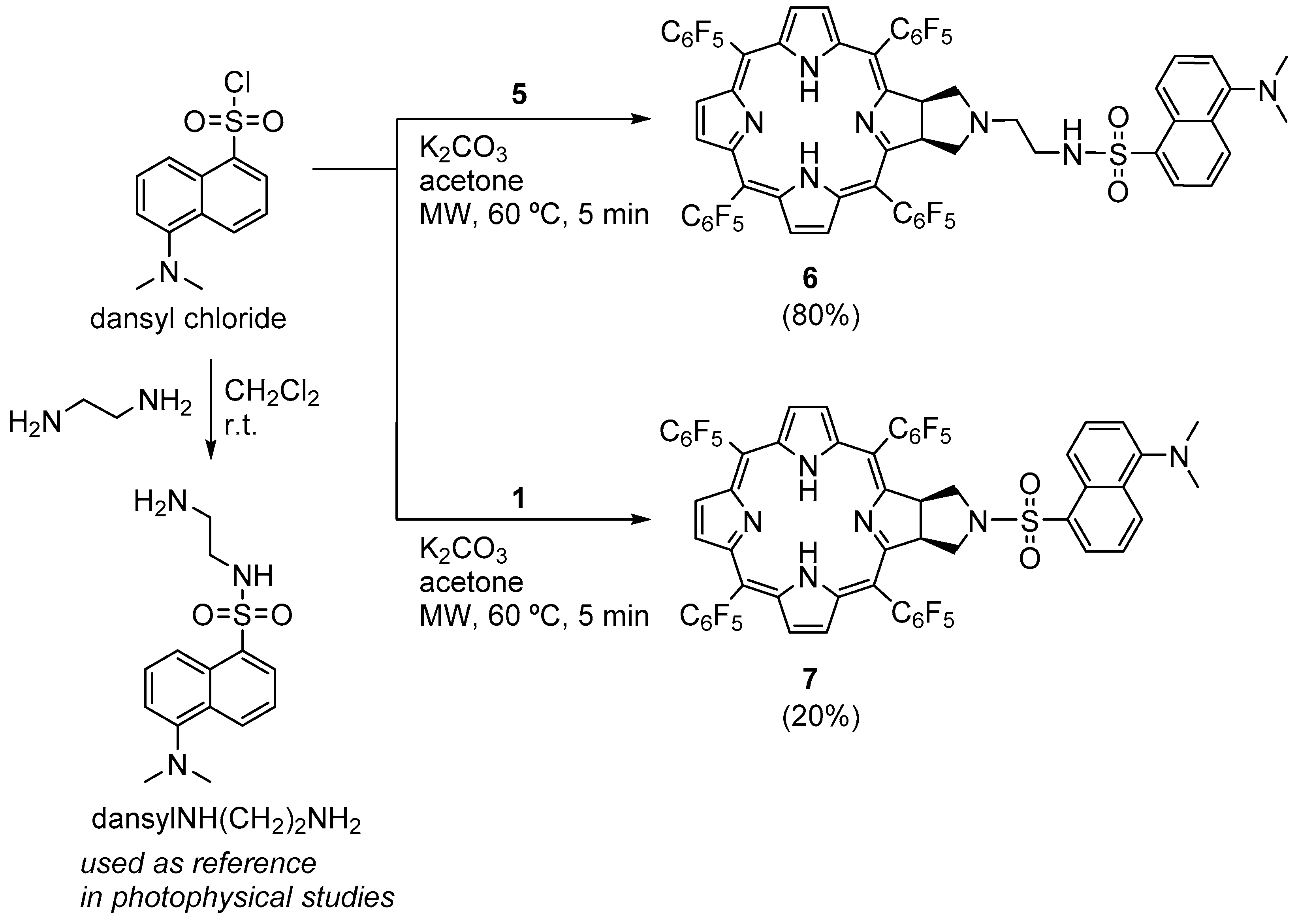
3.2.10. Synthesis of Chlorin−Dansyl Dyad 6
A solution of chlorin 5 (32 mg, 30 μmol) in acetone (2.5 mL) was transferred into a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. In a separate vial, a solution of dansyl chloride (17 mg, 64 μmol) in acetone (2.5 mL) was prepared and K2CO3 (41.2 mg, 300 μmol) was added. After thoroughly dissolving the dansyl chloride in the acetone solution, it was added to the chlorin 5 solution in the glass tube, sealed with a silicone septum, and placed into the microwave reactor cavity. The reaction mixture was then heated to 60 °C using a maximum microwave power of 30 W, which was automatically modulated for 5 min. After this time, the solution was diluted with 2.5 mL of dichloromethane and washed with deionized water and the organic extract was dried (Na2SO4), filtered, concentrated, and purified by silica gel column using CH2Cl2/MeOH (99:1) as the eluent, obtaining 31 mg of chlorin-dansyl 6 (80% yield). 1H NMR (400.14 MHz, CDCl3) δ (ppm): −1.83 (brs, 2H, NH), 2.29 (t, J = 5.5 Hz, 2H, N-CH2-CH2-NHSO2-), 2.44–2.48 (m, 2H, CH2-pyrrolidine), 2.85–2.90 (m, 8H: 6H, dansyl-N(CH3)2 + 2H, N-CH2-CH2-NHSO2-), 3.00–3.07 (m, 2H, CH2-pyrrolidine), 5.02 (t, J = 5.1 Hz, 1H, NH-sulfonamide), 5.05–5.08 (m, 2H, H-2 and H-3), 7.09 (d, J = 7.0, 1H, H-6′′-dansyl), 7.14–7.24 (m, 1H, H-7′′-dansyl), 7.47 (dd, J = 8.5 and 7.3 Hz, 1H, H-3′’-dansyl), 8.05 (d, J = 8.6 Hz, 1H, H-2′′-dansyl), 8.18 (dd, J = 7.3 and 1.2 Hz, 1H, H-8′′-dansyl), 8.36 (d, J = 4.9 Hz, 2H, H-β), 8.49 (s, 2H, H-β), 8.53 (d, J = 8.5 Hz, 1H, H-4′’-dansyl), 8.72 (d, J = 5.0 Hz, 2H, H-β). 19F{1H} NMR (376.46 MHz, CDCl3) δ −161.09 to −162.02 (m, 4F, Fmeta-Ar), −160.18 (td, 2F, J = 23.8 and 8.5 Hz, Fmeta-Ar), −159.78 (td, J = 23.8 and 8.4 Hz, 2F, Fmeta-Ar), −151.66 (t, J = 20.9 Hz, 2F, Fpara-Ar), −150.72 (t, J = 21.0 Hz, 2F, Fpara-Ar), −137.32 (dd, J = 24.1 and 8.0 Hz, 2F, Fortho-Ar), −136.76 to −137.07 (m, 2F, Fortho-Ar), −135.44 (dd, J = 24.1 and 7.5 Hz, 2F, Fortho-Ar); 13C{1H} NMR (100.63 MHz, CDCl3) δ (ppm): 41.10 (C-3′, N-CH2-CH2-NHSO2-), 45.31 (dansyl-N(CH3)2), 52.34 (C2-, C3-pyrrolidine), 53.30 (C2′-N-CH2-CH2-NHSO2), 60.22 (C21- C31-, CH2-pyrrolidine), 76.71, 77.02, 77.34, 96.74, 106.29, 115.00, 118.50, 123.02, 123.92, 127.90, 128.06, 129.59, 129.63, 129.89, 130.50, 132.45, 134.40, 135.31, 140.30, 152.06, 152.82, 168.35. UV-Vis (DMF) λmax (ε) 405 (144 × 103); 503 (13 × 103); 597 (4 × 103); 650 (37 × 103) nm. Fluorescence (DMF) λmax 655; 717 nm; ϕF = 0.188. HRMS (ESI) m/z: [M+H]+ Calcd for C60H32F20N7O2S+ 1294.201, Found 1294.192.
3.2.11. Synthesis of Chlorin−Dansyl Dyad 7
A solution of chlorin 1 (16 mg, 16 μmol) in acetone (2.5 mL) was transferred to a 10 mL thick-walled glass tube equipped with a Teflon-coated magnetic stir bar. In a separate vial, a solution of dansyl chloride (9.0 mg, 33 μmol) in acetone (2.5 mL) was prepared and K2CO3 (22.1 mg, 160 μmol) was added. After thoroughly dissolving the dansyl chloride, the solution was added to the chlorin 1 solution, the glass tube was sealed with a silicone septum and placed into the microwave reactor cavity. The reaction mixture was then heated to 60 °C using a maximum microwave power of 30 W, which was automatically modulated for 5 min. After this time, the solution was diluted with dichloromethane (2.5 mL) and washed with deionized water and the organic extract was dried (Na2SO4), filtered, concentrated, and purified by silica gel column using CH2Cl2/acetone (98:2) as the eluent. Four mg of chlorin−dansyl 7 (20% yield) were obtained. 1H NMR (400.14 MHz, CDCl3) δ (ppm): −2.15 (s, 2H, NH), 2.20 (s, 6H, dansyl-N(CH3)2), 3.28 (dd, J = 10.3 and 4.5 Hz, 2H, CH2-pyrrolidine), 3.89–3.98 (m, 2H, CH2-pyrrolidine), 5.12–5.24 (m, 2H, H-2,3), 6.08 (dd, J = 7.6 and 0.9 Hz, 1H, H-6′′-dansyl), 6.33 (dd, J = 8.7 and 7.6 Hz, 1H, H-7′′-dansyl), 7.20 (t, J = 8.0 Hz, 1H, H-3′′-dansyl), 7.77 (d, J = 8.7 Hz, 1H, H-2′′-dansyl), 7.97 (d, J = 7.7 Hz, 2H, H-8′′ and H-4′′-dansyl), 8.31 (d, J = 5.0 Hz, 2H, H-β), 8.48 (s, 2H, H-β), 8.68 (d, J = 5.0 Hz, 2H, H-β). 19F{1H} NMR (376.46 MHz; CDCl3) δ (ppm): −161.48 to −161.13 (m, 4F, Fmeta-Ar), −159.33 (dtd, J = 53.8, 22.5 and 8.5 Hz, 4F, Fmeta-Ar), −151.38 (t, J = 20.8 Hz, 2F, Fpara-Ar), −150.00 (t, J = 20.9 Hz, 2F, Fpara-Ar), −137.28 (dd, J = 23.9 and 8.7 Hz, 4F, Fortho-Ar), −137.12 to −136.84 (m, 2F, Fortho-Ar) −135.15 (dd, J = 24.7 and 8.5 Hz, 2F, Fortho-Ar); 13C{1H} NMR (100.63 MHz, CDCl3) δ (ppm): 44.73, 52.30, 54.09, 96.70, 114.15, 118.14, 122.48, 124.13, 127.33, 128.27, 129.25, 129.99, 130.50, 130.91, 131.31, 132.78, 135.43, 140.29, 151.00, 153.09, 166.76. UV-Vis (DMF) λmax (ε) 405 (132 × 103); 502 (11 × 103); 597 (4 × 103); 650 (35 × 103) nm. Fluorescence (DMF) λmax 655; 716 nm; ϕF = 0.123. HRMS (ESI) m/z: [M+H]+ Calcd for C58H27F20N6O2S+ 1251.159, Found 1251.161.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}