
Topoisomerase Inhibitors and PIM1 Kinase Inhibitors Improve Gene Editing Efficiency Mediated by CRISPR-Cas9 and Homology-Directed Repair


Abstract
:1. Introduction
2. Results
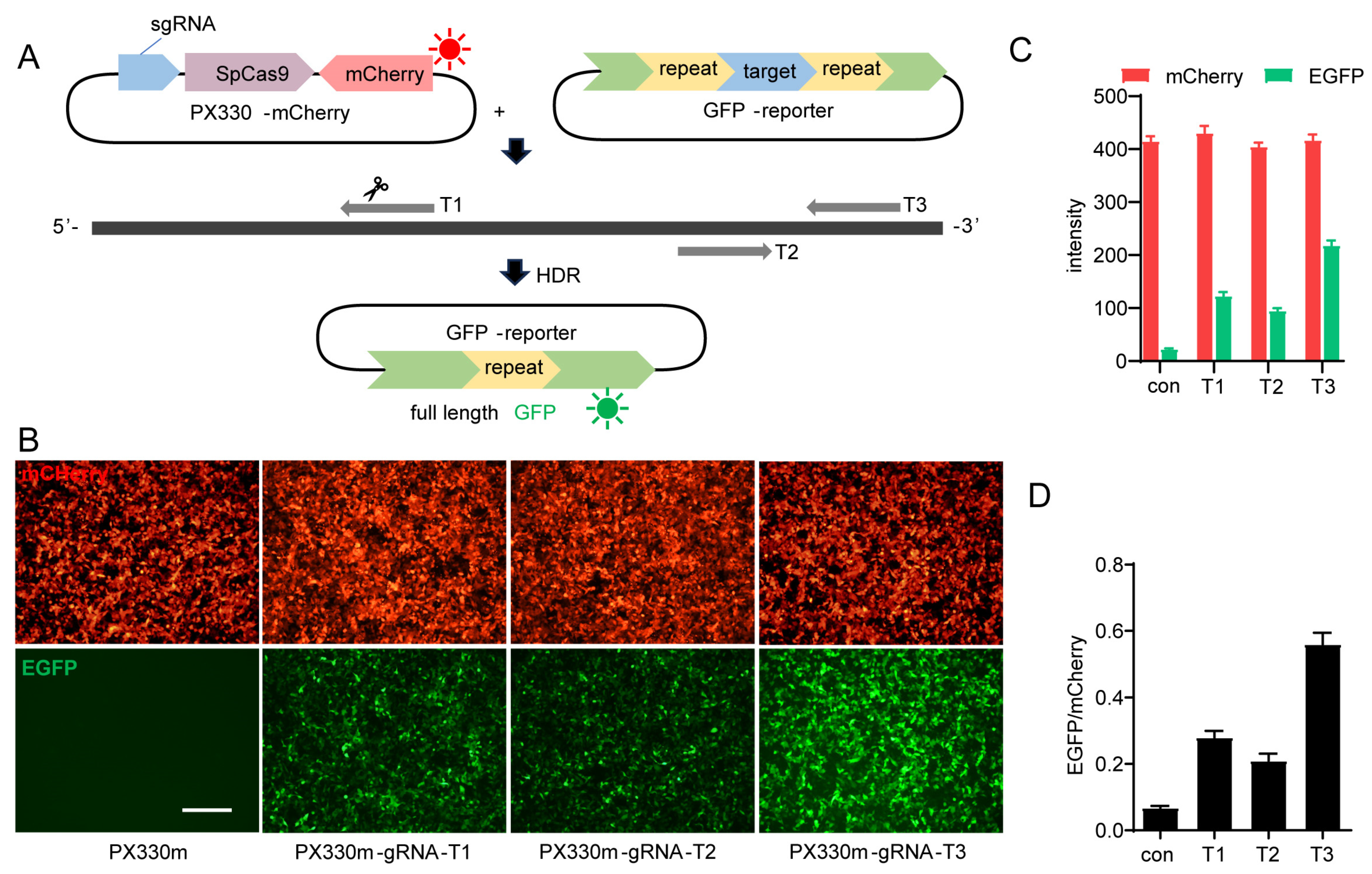
2.1. Design of the In Vitro GFP Reporter-Based HDR Model
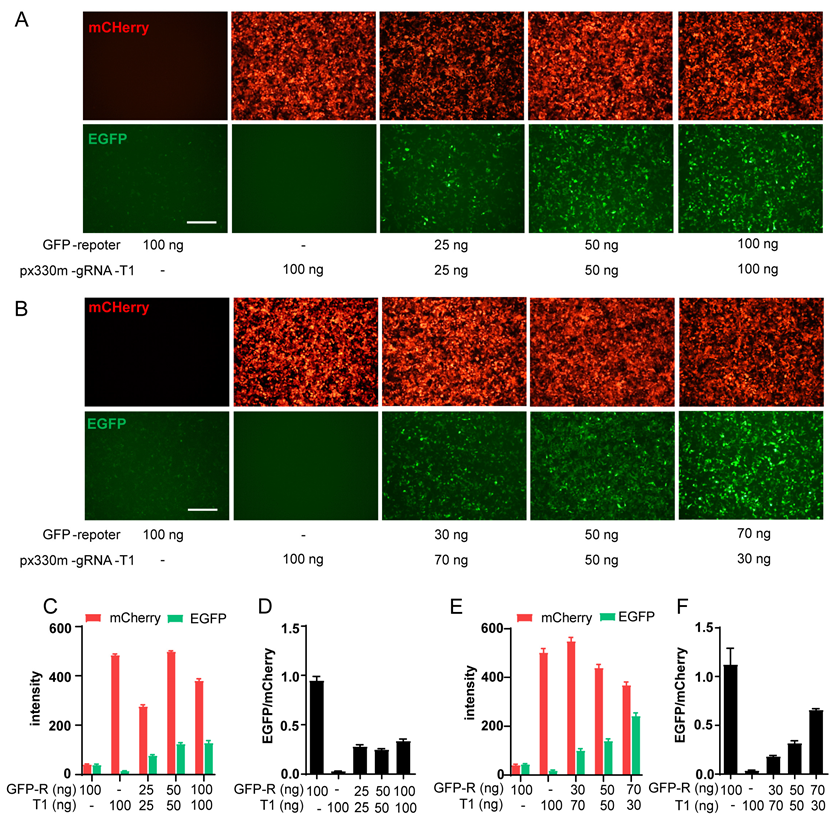
2.2. Optimization of GFP-Report Detection Model and Compound Screening
2.3. Enhanced Efficiency of HDR in the Reporter System by Topoisomerase Inhibitors and PIM1 Kinase Inhibitors
2.4. Etoposide and Quercetagetin Enhance CRISPR/HDR-Mediated Gene Knock-in in Mouse Zygotes
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction
4.2. Cell Culture and Transfections
4.3. Compound Screening
4.4. Embryonic Genomic DNA Extraction and PCR Validation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, S.K.; Shukla, P. Gene editing for cell engineering: Trends and applications. Crit. Rev. Biotechnol. 2017, 37, 672–684. [Google Scholar] [CrossRef]
- Lu, X.-J.; Xue, H.-Y.; Ke, Z.-P.; Chen, J.-L.; Ji, L.-J. CRISPR-Cas9: A new and promising player in gene therapy. J. Med. Genet. 2015, 52, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; et al. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 2019, 232, 116636. [Google Scholar] [CrossRef] [PubMed]
- Rengasamy, B.; Manna, M.; Thajuddin, N.B.; Sathiyabama, M.; Sinha, A.K. Breeding rice for yield improvement through CRISPR/Cas9 genome editing method: Current technologies and examples. Physiol. Mol. Biol. Plants 2024, 30, 185–198. [Google Scholar] [CrossRef]
- Liang, Y.; Gao, S.; Qi, X.; Valentovich, L.N.; An, Y. Progress in Gene Editing and Metabolic Regulation of Saccharomyces cerevisiae with CRISPR/Cas9 Tools. ACS Synth. Biol. 2024, 13, 428–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cottle, W.T.; Ha, T. Mapping cellular responses to DNA double-strand breaks using CRISPR technologies. Trends Genet. 2023, 39, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Danner, E.; Bashir, S.; Yumlu, S.; Wurst, W.; Wefers, B.; Kuhn, R. Control of gene editing by manipulation of DNA repair mechanisms. Mamm. Genome 2017, 28, 262–274. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, S.; Hur, J.K. CRISPR and Target-Specific DNA Endonucleases for Efficient DNA Knock-in in Eukaryotic Genomes. Mol. Cells 2018, 41, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2018, 9, 691. [Google Scholar] [CrossRef]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, K.; Kunihiro, Y.; Kaneko, T.; Nagahora, H.; Voigt, B.; Mashimo, T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016, 7, 10431. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, M.; Choudhary, B.; Raghavan, S.C. SCR7, a potent cancer therapeutic agent and a biochemical inhibitor of nonhomologous DNA end-joining. Cancer Rep. 2021, 4, e1341. [Google Scholar] [CrossRef] [PubMed]
- Lamas-Toranzo, I.; Martinez-Moro, A.; O’Callaghan, E.; Millan-Blanca, G.; Sanchez, J.M.; Lonergan, P.; Bermejo-Alvarez, P. RS-1 enhances CRISPR-mediated targeted knock-in in bovine embryos. Mol. Reprod. Dev. 2020, 87, 542–549. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Yang, Z.X.; Zhang, F.; Fu, Y.W.; Dai, X.Y.; Wen, W.; Zhang, B.; Choi, H.; Chen, W.; Brown, M.; et al. HDAC inhibitors improve CRISPR-mediated HDR editing efficiency in iPSCs. Sci. China Life Sci. 2021, 64, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.E.; Matsumoto, Y.; Brooks, R.C.; Lu, Z.; Lieber, M.R.; Tomkinson, A.E. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair 2016, 43, 18–23. [Google Scholar] [CrossRef]
- Xie, Z.; Pang, D.; Wang, K.; Li, M.; Guo, N.; Yuan, H.; Li, J.; Zou, X.; Jiao, H.; Ouyang, H.; et al. Optimization of a CRISPR/Cas9-mediated Knock-in Strategy at the Porcine Rosa26 Locus in Porcine Foetal Fibroblasts. Sci. Rep. 2017, 7, 3036. [Google Scholar] [CrossRef]
- Ray, U.; Gopinatha, V.K.; Sharma, S.; Goyary, L.; Choudhary, B.; Mantelingu, K.; Rangappa, K.S.; Raghavan, S.C. Identification and characterization of mercaptopyrimidine-based small molecules as inhibitors of nonhomologous DNA end joining. FEBS J. 2023, 290, 796–820. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Zhang, C.; Guo, S.; Liang, X.; Lan, Y.; Wang, M.; Zhuo, N.; Yin, J.; Liu, H.; Gu, M.; et al. Identification of Betulinic Acid Derivatives as Potent TGR5 Agonists with Antidiabetic Effects via Humanized TGR5(H88Y) Mutant Mice. J. Med. Chem. 2021, 64, 12181–12199. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Saha, S.; Wang, W.; Saha, L.K.; Huang, S.N.; Pommier, Y. Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair 2020, 89, 102837. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Gao, C. Topoisomerase Inhibitors and Targeted Delivery in Cancer Therapy. Curr. Top. Med. Chem. 2019, 19, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Berger, J.M. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes 2019, 10, 859. [Google Scholar] [CrossRef] [PubMed]
- Darcy, N.; Gabrielli, B. Topoisomerase II Inhibitors and Poisons, and the Influence of Cell Cycle Checkpoints. Curr. Med. Chem. 2017, 24, 1504–1519. [Google Scholar] [CrossRef]
- Zhang, X.; Song, M.; Kundu, J.K.; Lee, M.H.; Liu, Z.Z. PIM Kinase as an Executional Target in Cancer. J. Cancer Prev. 2018, 23, 109–116. [Google Scholar] [CrossRef]
- Toth, R.K.; Warfel, N.A. Targeting PIM Kinases to Overcome Therapeutic Resistance in Cancer. Mol. Cancer Ther. 2021, 20, 3–10. [Google Scholar] [CrossRef]
- Santio, N.M.; Koskinen, P.J. PIM kinases: From survival factors to regulators of cell motility. Int. J. Biochem. Cell Biol. 2017, 93, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PIM kinase inhibitors: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. [Google Scholar] [CrossRef]
- Morishita, D.; Katayama, R.; Sekimizu, K.; Tsuruo, T.; Fujita, N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008, 68, 5076–5085. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Nucleotide Sequence |
|---|---|
| sgRNA-T1 | CTATGGAGCCGGAACCATCA |
| sgRNA-T2 | CAGCAGCAGATTGGCAAGCA |
| sgRNA-T3 | ACCAGTAGCCCTGATGGTTC |
| Target sequence in GFP reporter model | TGGGCTATGGAGCCGGAACCATCAGGGCTACTGGTCCTGCCTCCTTCTCCACTTGACCCCCAACTTTTGTTTCCTTTCCCTGCTTGCCAATCTGCTGCTG |
| ssODN for zygote injection | TCACAGGGCTGGCACTGCCCATGCTGCCTGGGCTATGGAGCCGGAACCATCCATAAGGATCCACGACGTCCAGCGCTACTGGTCCTGCCTCCTTCTCCACTTGACCCCCAACTTTTGTTT |
| Forward primer for nested PCR (long) | ACTGAGCTGTCGGCCATTCC |
| Reverse primer for nested PCR (long) | AGACAGCTTGGGAGCTGCAG |
| Forward primer for nested PCR (short) | TAGCCGGGCTGCTCACAGGG |
| Reverse primer for nested PCR (short) | CAAGCAGGGAAAGGAAACAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, Y.; Wang, M.; Guo, S.; Xie, X. Topoisomerase Inhibitors and PIM1 Kinase Inhibitors Improve Gene Editing Efficiency Mediated by CRISPR-Cas9 and Homology-Directed Repair. Molecules 2024, 29, 2890. https://doi.org/10.3390/molecules29122890
Yun Y, Wang M, Guo S, Xie X. Topoisomerase Inhibitors and PIM1 Kinase Inhibitors Improve Gene Editing Efficiency Mediated by CRISPR-Cas9 and Homology-Directed Repair. Molecules. 2024; 29(12):2890. https://doi.org/10.3390/molecules29122890
Chicago/Turabian StyleYun, Ying, Min Wang, Shimeng Guo, and Xin Xie. 2024. "Topoisomerase Inhibitors and PIM1 Kinase Inhibitors Improve Gene Editing Efficiency Mediated by CRISPR-Cas9 and Homology-Directed Repair" Molecules 29, no. 12: 2890. https://doi.org/10.3390/molecules29122890