Boosting the Electrocatalytic Oxygen Reduction Activity of MnN4-Doped Graphene by Axial Halogen Ligand Modification


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
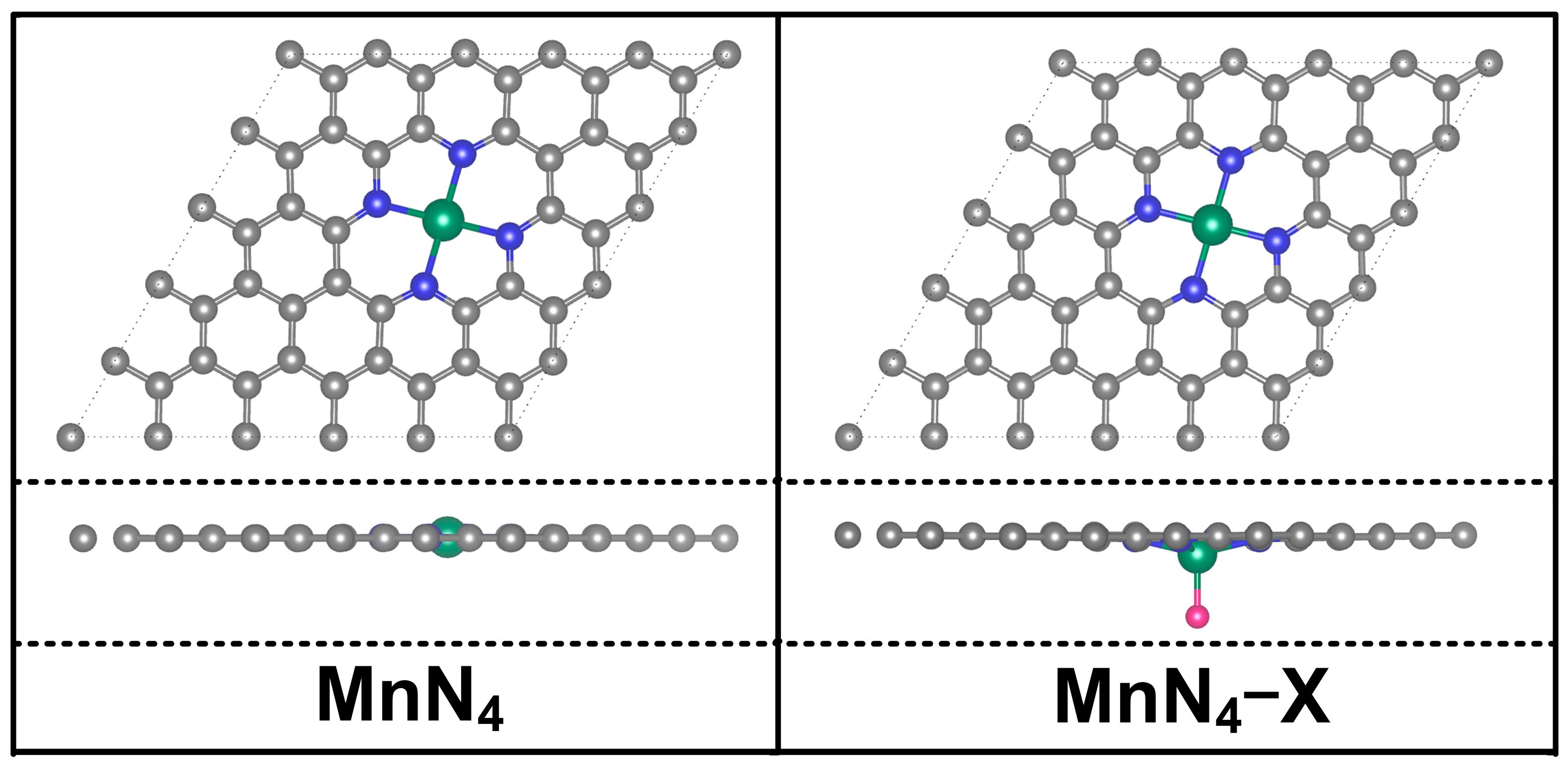
2.1. Structural Model
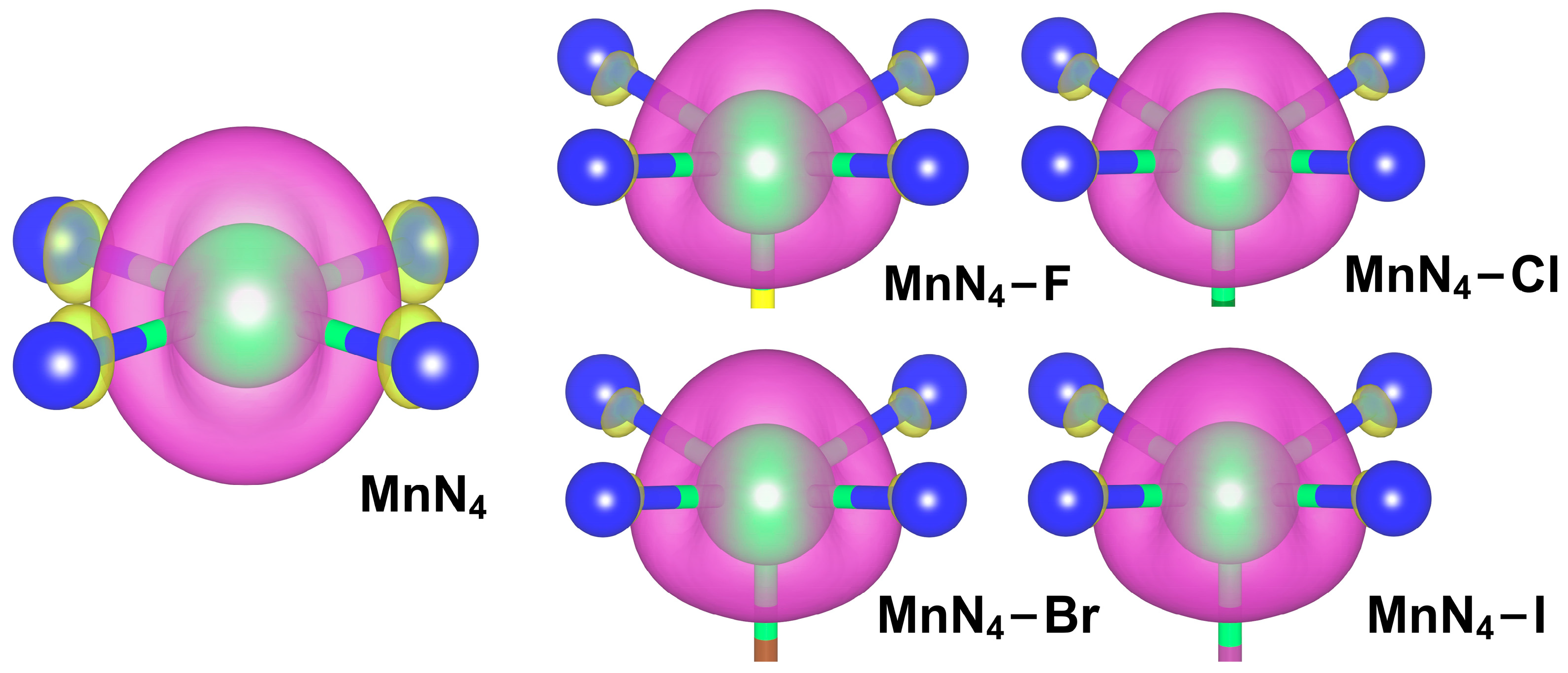
2.2. Adsorption of Intermediates
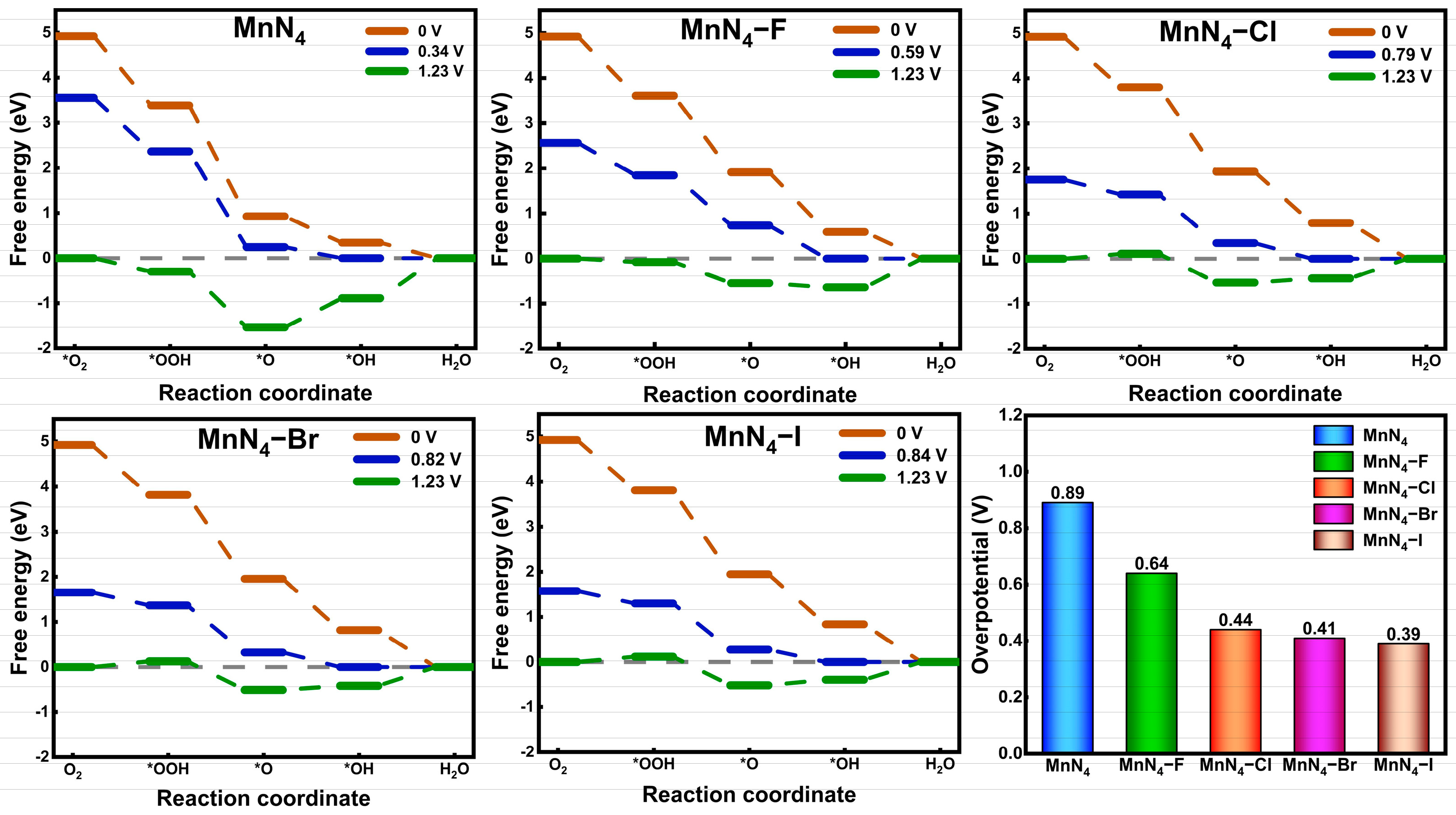
2.3. Catalytic Performance
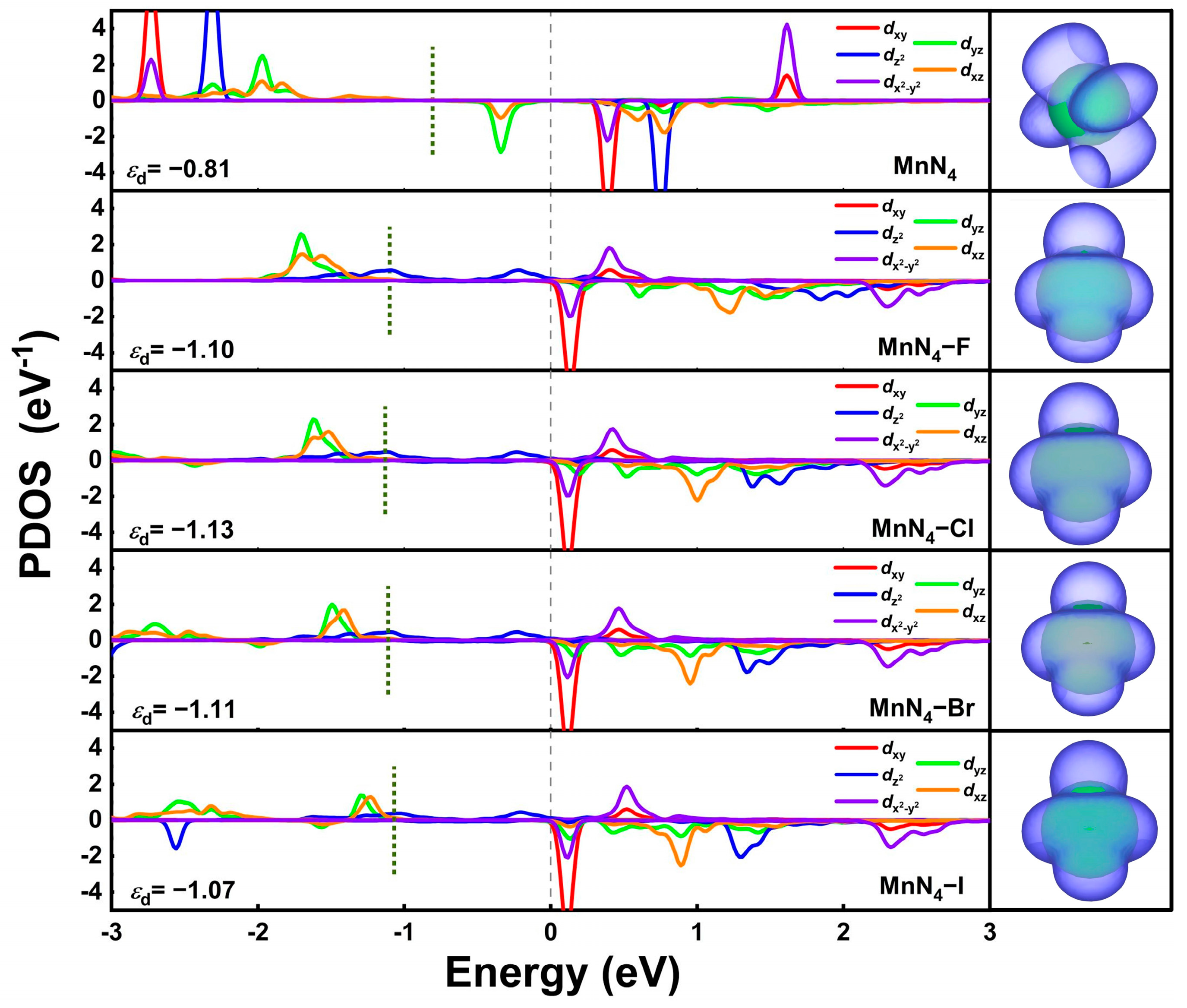
2.4. Origin of the Catalytic Activity
3. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, H.F.; Xu, Q. Materials design for rechargeable metal-air batteries. Matter 2019, 1, 565–595. [Google Scholar] [CrossRef]
- Zhang, Q.N.; Dong, S.D.; Shao, P.P.; Zhu, Y.H.; Mu, Z.J.; Sheng, D.F.; Zhang, T.; Jiang, X.; Shao, R.W.; Ren, Z.; et al. Covalent organic framework-based porous ionomers for high-performance fuel cells. Science 2022, 378, 181–186. [Google Scholar] [CrossRef]
- Brett, D.J.L.; Aguiar, P.; Brandon, N.P.; Bull, R.N.; Galloway, R.C.; Hayes, G.W.; Lillie, K.; Mellors, C.; Millward, M.; Smith, C.; et al. Project absolute: A zebra battery/Intermediate temperature solid oxide fuel cell hybrid for automotive applications. J. Fuel Cell Sci. Technol. 2006, 3, 254–262. [Google Scholar] [CrossRef]
- Mukerjee, S.; Srinivasan, S.; Soriaga, M.P.; Mcbreen, J. Role of structural and electronic properties of Pt and Pt alloys on electrocatalysis of oxygen reduction: An in situ XANES and EXAFS investigation. J. Electrochem. Soc. 1995, 142, 1409–1422. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Origin of the unique activity of Pt/TiO2 catalysts for the water–gas shift reaction. J. Catal. 2013, 306, 78–90. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Huang, L.; Zaman, S.; Tian, X.L.; Wang, Z.T.; Fang, W.S.; Xia, B.Y. Advanced platinum-based oxygen reduction electrocatalysts for fuel cells. Acc. Chem. Res. 2021, 54, 311–322. [Google Scholar] [CrossRef]
- Niu, W.J.; He, J.Z.; Gu, B.N.; Liu, M.C.; Chueh, Y.L. Opportunities and challenges in precise synthesis of transition metal single-atom supported by 2D materials as catalysts toward oxygen reduction reaction. Adv. Funct. Mater. 2021, 31, 2103558. [Google Scholar] [CrossRef]
- Li, L.; Huang, R.; Cao, X.R.; Wen, Y.H. Computational screening of efficient graphene-supported transition metal single atom catalysts toward the oxygen reduction reaction. J. Mater. Chem. A 2020, 8, 19319–19327. [Google Scholar] [CrossRef]
- Zhuo, H.Y.; Zhang, X.; Liang, J.X.; Yu, Q.; Xiao, H.; Li, J. Theoretical understandings of graphene-based metal single-atom catalysts: Stability and catalytic performance. Chem. Rev. 2020, 120, 12315–12341. [Google Scholar] [CrossRef]
- Wang, J.; Liu, W.; Luo, G.; Li, Z.; Zhao, C.; Zhang, H.R.; Zhu, M.; Xu, Q.; Wang, X.Q.; Zhao, C.M.; et al. Synergistic effect of well-defined dual sites boosting the oxygen reduction reaction. Energy Environ. Sci. 2018, 11, 3375–3379. [Google Scholar] [CrossRef]
- Luo, E.G.; Chu, Y.Y.; Liu, J.; Shi, Z.P.; Zhu, S.Y.; Gong, L.Y.; Ge, J.J.; Choi, C.H.; Liu, C.P.; Xing, W. Pyrolyzed M–Nx catalysts for oxygen reduction reaction: Progress and prospects. Energy Environ. Sci. 2021, 14, 2158–2185. [Google Scholar] [CrossRef]
- Zhou, Y.; Xing, Y.F.; Wen, J.; Ma, H.B.; Wang, F.B.; Xia, X.H. Axial ligands tailoring the ORR activity of cobalt porphyrin. Sci. Bull. 2019, 64, 1158–1166. [Google Scholar] [CrossRef]
- Sun, J.K.; Pan, Y.W.; Xu, M.Q.; Sun, L.; Zhang, S.L.; Deng, W.Q.; Zhai, D. Heteroatom doping regulates the catalytic performance of single-atom catalyst supported on graphene for ORR. Nano Res. 2024, 17, 1086–1093. [Google Scholar] [CrossRef]
- Li, R.Z.; Wang, D.S. Superiority of dual-atom catalysts in electrocatalysis: One step further than single-stom catalysts. Adv. Energy Mater. 2022, 12, 2103564. [Google Scholar] [CrossRef]
- Wang, G.L.; Yang, X.L.; Wang, R.Y.; Jia, J.F. A theoretical study on the ORR electrocatalytic activity of axial ligand modified cobalt polyphthalocyanine. Phys. Chem. Chem. Phys. 2023, 25, 27342–27351. [Google Scholar] [CrossRef]
- She, X.L.; Gao, J.H.; Gao, Y.; Tang, H.; Li, K.; Wang, Y.; Wu, Z.J. Axial ligand engineering for highly efficient oxygen reduction catalysts in transition metal–N4 doped graphene. New J. Chem. 2022, 46, 16138–16150. [Google Scholar] [CrossRef]
- Shakir, R.; Komsa, H.P.; Sinha, A.S.K.; Karthikeyan, J. Electronic origin of enhanced selectivity through the halogenation of a single Mn atom on graphitic C3N4 for electrocatalytic reduction of CO2 from first-principles calculations. J. Phys. Chem. C 2023, 127, 14694–14703. [Google Scholar] [CrossRef]
- Liu, X.J.; Liu, Y.R.; Yang, W.X.; Feng, X.; Wang, B. Controlled modification of axial coordination for transition-metal single-atom electrocatalyst. Chem.-A Eur. J. 2022, 28, e202201471. [Google Scholar] [CrossRef]
- Liu, Y.N.; Wang, D.S.; Yang, B.; Li, Z.J.; Peng, X.Y.; Liu, Z.B.; Zeng, L.B.; Zhang, T.; Rodriguez, R.; Lei, L.; et al. Efficiently electrochemical CO2 reduction on molybdenum-nitrogen-carbon catalysts with optimized p-block axial ligands. Chem. Eng. Sci. 2023, 273, 118638. [Google Scholar] [CrossRef]
- Nagaprasad Reddy, S.; Krishnamurthy, C.B.; Grinberg, I. First-principles study of the ligand substituent effect on ORR catalysis by metallocorroles. J. Phys. Chem. C 2020, 124, 11275–11283. [Google Scholar] [CrossRef]
- Han, Y.H.; Wang, Y.G.; Xu, R.R.; Chen, W.X.; Zheng, L.R.; Han, A.J.; Zhu, Y.Q.; Zhang, J.; Zhang, H.B.; Luo, J.; et al. Electronic structure engineering to boost oxygen reduction activity by controlling the coordination of the central metal. Energy Environ. Sci. 2018, 11, 2348–2352. [Google Scholar] [CrossRef]
- Zhao, K.M.; Liu, S.Q.; Li, Y.Y.; Wei, X.L.; Ye, G.Y.; Zhu, W.W.; Su, Y.K.; Wang, J.; Liu, H.T.; He, Z.; et al. Insight into the mechanism of axial ligands regulating the catalytic activity of Fe–N4 sites for oxygen reduction reaction. Adv. Energy Mater. 2022, 12, 2103588. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.Z.; Zhao, X.; Yu, H.; Zhang, H. Effect of the axial halogen ligand on the oxygen reduction reaction performance of transition metal–nitrogen–carbon catalysts. J. Phys. Chem. C 2023, 127, 14107–14116. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Bian, X.Q.; Xie, S.Y.; Ruan, W.Q.; Chen, W.K.; Guo, X.Y.; Ding, K.N. Axial halogen coordinated metal-nitrogen-carbon moiety enables efficient electrochemical oxygen reduction to hydrogen peroxide. Int. J. Hydrog. Energy 2024, 51, 1413–1420. [Google Scholar] [CrossRef]
- Li, L.; Li, Y.M.; Huang, R.; Cao, X.R.; Wen, Y.H. Single Mn atom anchored on nitrogen-doped graphene as a highly efficient electrocatalyst for oxygen reduction reaction. Chem. Eur. J. 2021, 27, 9686–9693. [Google Scholar] [CrossRef]
- Duan, Z.Y.; Wang, G.F. Comparison of reaction energetics for oxygen reduction reactions on Pt(100), Pt(111), Pt/Ni(100), and Pt/Ni(111) surfaces: A first-principles study. J. Phys. Chem. C 2013, 117, 6284–6292. [Google Scholar] [CrossRef]
- Tripković, V.; Skúlason, E.; Siahrostami, S.; Nørskov, J.K.; Rossmeisl, J. The oxygen reduction reaction mechanism on Pt(111) from density functional theory calculations. Electrochim. Acta 2010, 55, 7975–7981. [Google Scholar] [CrossRef]
- Orellana, W. Catalytic properties of transition metal–N4 moieties in graphene for the oxygen reduction reaction: Evidence of spin-dependent mechanisms. J. Phys. Chem. C 2013, 117, 9812–9818. [Google Scholar] [CrossRef]
- Lu, Z.S.; Xu, G.L.; He, C.Z.; Wang, T.X.; Yang, L.; Yang, Z.X.; Ma, D.W. Novel catalytic activity for oxygen reduction reaction on MnN4 embedded graphene: A dispersion-corrected density functional theory study. Carbon 2015, 84, 500–508. [Google Scholar] [CrossRef]
- Xu, H.X.; Cheng, D.J.; Cao, D.P.; Zeng, X.C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Gerber, I.C.; Serp, P. A theory/experience description of support effects in carbon-supported catalysts. Chem. Rev. 2020, 120, 1250–1349. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.Q.; Zhong, J.; Xu, H.Y.; Huang, Y.C.; Hu, Z.W.; Chen, B.; Zhang, Y.; Ni, M.; Xu, X.M.; Zhou, W.; et al. A universal chemical-induced tensile strain tuning strategy to boost oxygen-evolving electrocatalysis on perovskite oxides. Appl. Phys. Rev. 2022, 9, 011422. [Google Scholar] [CrossRef]
- Li, L.; Wu, X.X.; Du, Q.Y.; Bai, N.S.; Wen, Y.H. Boosting the oxygen reduction reaction activity of dual-atom catalysts on N-doped graphene by regulating the N coordination environment. Phys. Chem. Chem. Phys. 2024, 26, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Y.; Huang, R.; Cao, X.R.; Wen, Y.H. Boosting the electrocatalytic activity of Fe−Co dual-atom catalysts for oxygen reduction reaction by ligand-modification engineering. ChemCatChem 2021, 13, 4645–4651. [Google Scholar] [CrossRef]
- Guo, X.Y.; Lin, S.R.; Gu, J.X.; Zhang, S.L.; Chen, Z.F.; Huang, S.P. Simultaneously achieving high activity and selectivity toward two-electron O2 electroreduction: The power of single-atom catalysts. ACS Catal. 2019, 9, 11042–11054. [Google Scholar] [CrossRef]
- Zhao, X.H.; Liu, Y.Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 2021, 143, 9423–9428. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.Q.; Zhou, J.; Huang, Y.C.; Dong, C.L.; Wang, J.Q.; Zhou, W.; Shao, Z.P. Screening highly active perovskites for hydrogen-evolving reaction via unifying ionic electronegativity descriptor. Nat. Commun. 2019, 10, 3755. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B-Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurateab initioparametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Nørskov, J.K. Electrochemical dissolution of surface alloys in acids: Thermodynamic trends from first-principles calculations. Electrochim. Acta 2007, 52, 5829–5836. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, S.; Zhao, R.; Yu, W.; Li, L.; Zhang, M. Boosting the Electrocatalytic Oxygen Reduction Activity of MnN4-Doped Graphene by Axial Halogen Ligand Modification. Molecules 2024, 29, 3517. https://doi.org/10.3390/molecules29153517
Wei S, Zhao R, Yu W, Li L, Zhang M. Boosting the Electrocatalytic Oxygen Reduction Activity of MnN4-Doped Graphene by Axial Halogen Ligand Modification. Molecules. 2024; 29(15):3517. https://doi.org/10.3390/molecules29153517
Chicago/Turabian StyleWei, Shaoqiang, Ran Zhao, Wenbo Yu, Lei Li, and Min Zhang. 2024. "Boosting the Electrocatalytic Oxygen Reduction Activity of MnN4-Doped Graphene by Axial Halogen Ligand Modification" Molecules 29, no. 15: 3517. https://doi.org/10.3390/molecules29153517