

Why Does the Optimal Tuning Method of the Range Separation Parameter of a Long-Range Corrected Density Functional Fail in Intramolecular Charge Transfer Excitation Calculations?


Abstract
:1. Introduction
2. Results and Discussion
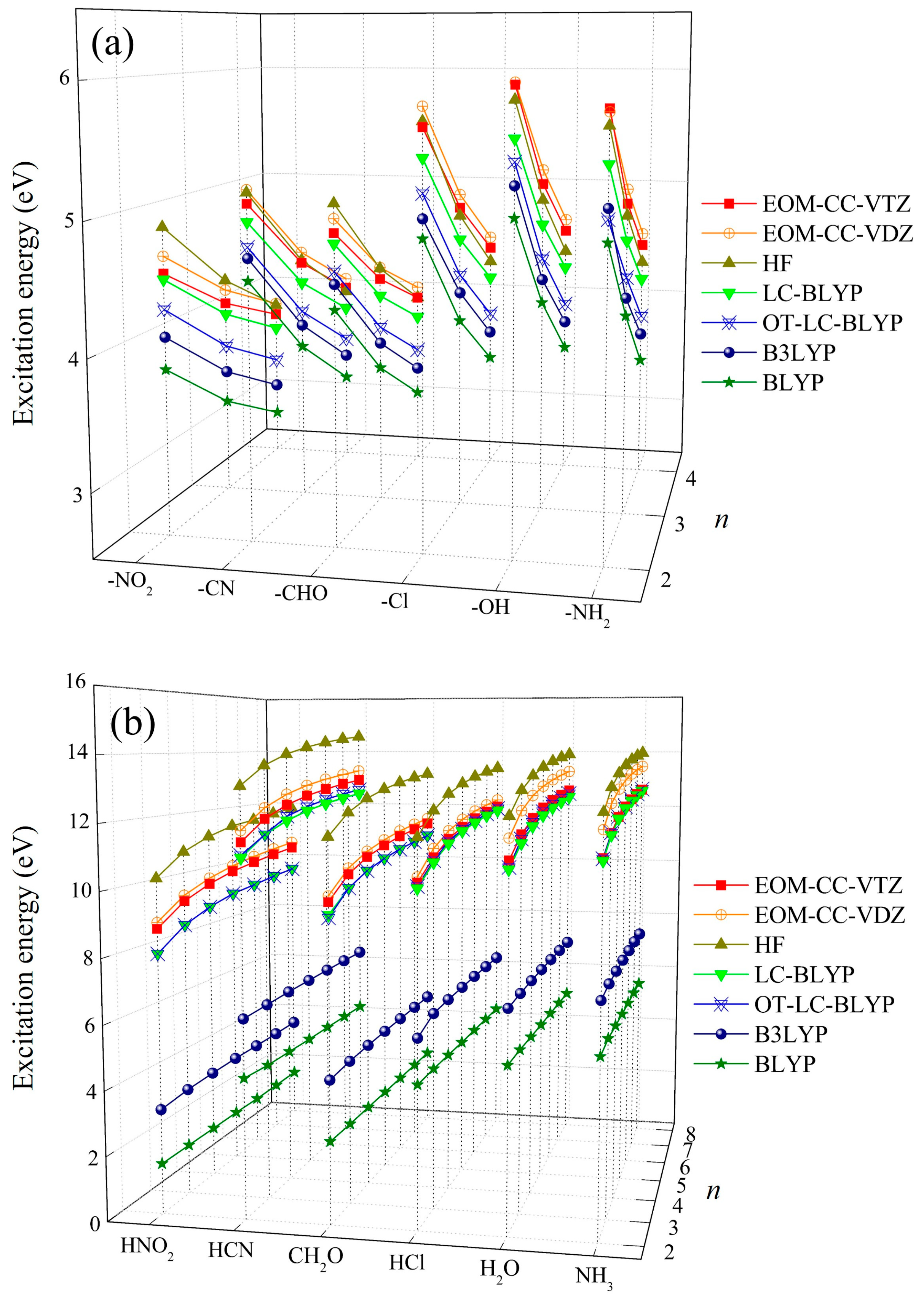
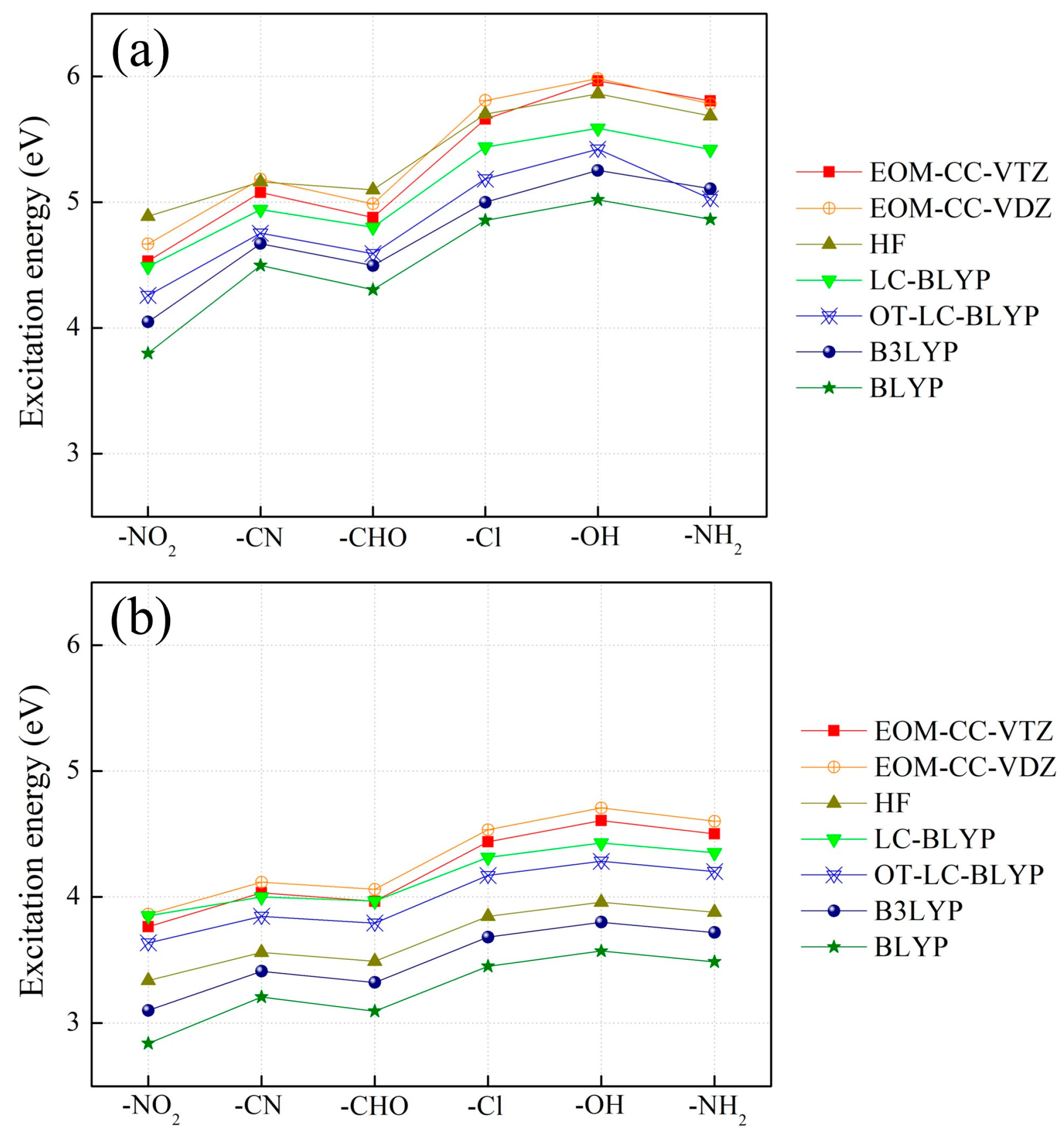
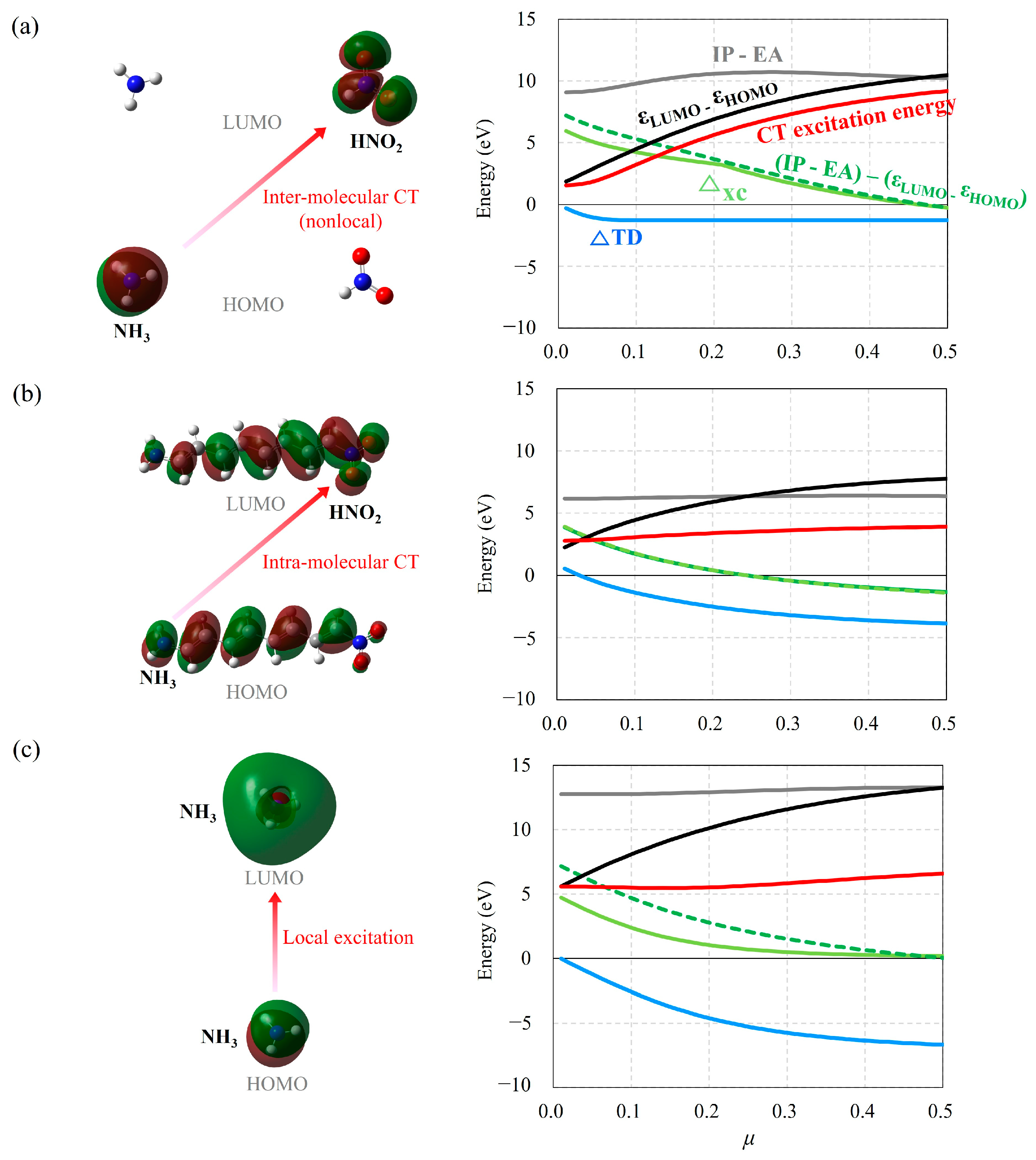
2.1. The Effect of Electron Acceptors on Intra- and Intermolecular CT Excitation Energies
2.2. Assessment of Various DFT Functionals on Intra- and Intermolecular CT Excitation Energy
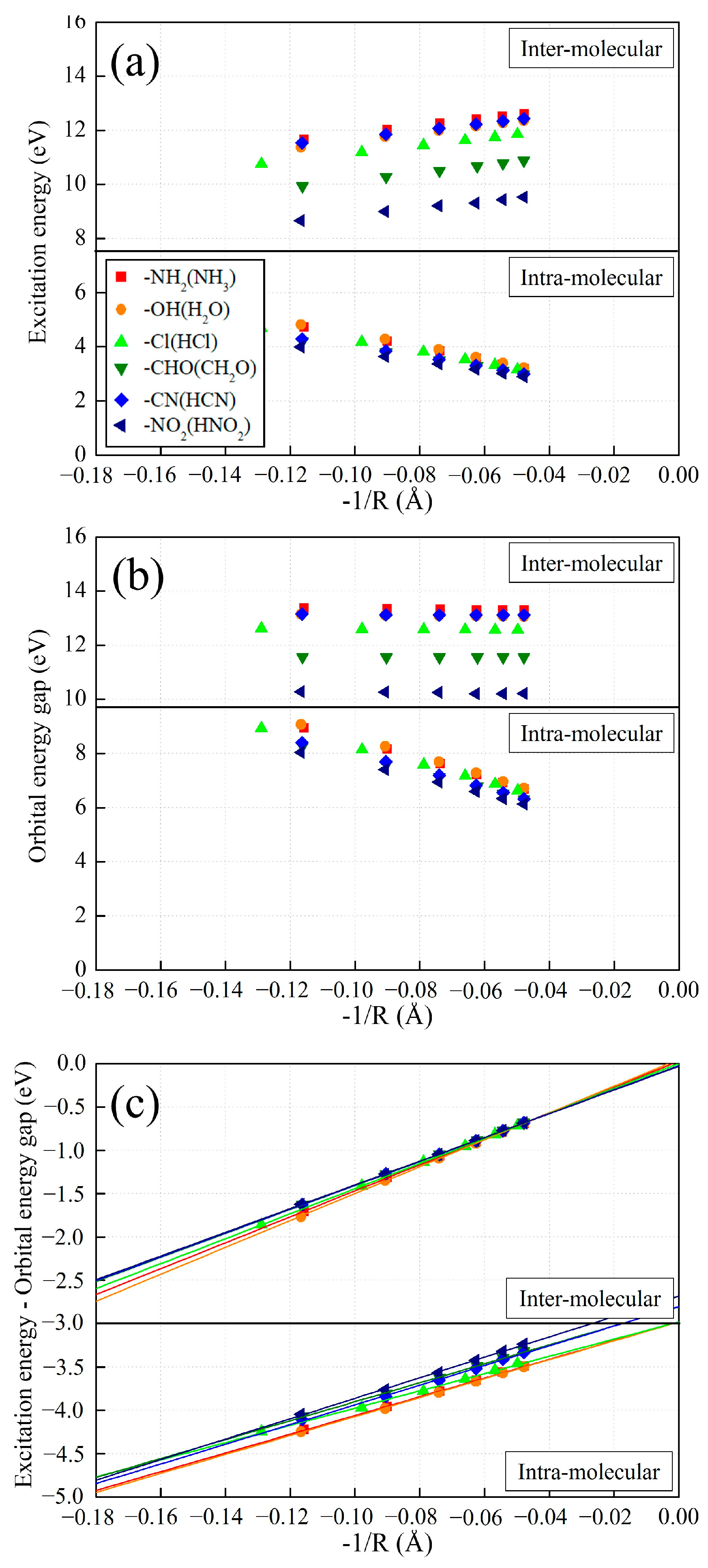
2.3. Exciton Binding Energy in Intra- and Intermolecular CT Excitation
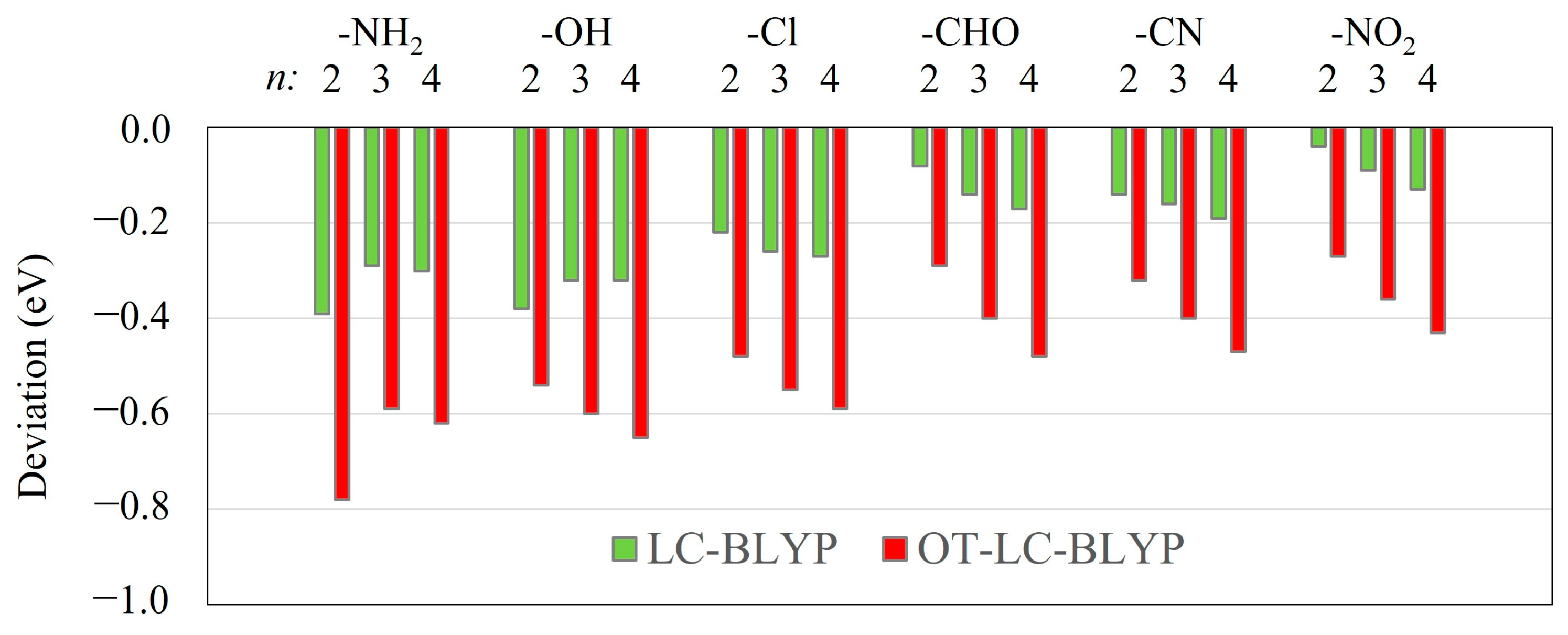
2.4. Ineffectiveness of Optimal Tuning Method in Intramolecular CT Excitation Calculations
3. Theory
3.1. Time-Dependent Density Functional Theory with Hybrid- and LC-DFTs
3.2. Asymptotic Potential and Excitonic Bindng Energy
3.3. Derivative Discontinuity of Approximated Exchange–Correlation Functionals
4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Minami, T.; Nakano, M.; Castet, F. Nonempirically Tuned Long-Range Corrected Density Functional Theory Study on Local and Charge-Transfer Excitation Energies in a Pentacene/C-60 Model Complex. J. Phys. Chem. Lett. 2011, 2, 1725–1730. [Google Scholar] [CrossRef]
- Zhang, C.R.; Sears, J.S.; Yang, B.; Aziz, S.G.; Coropceanu, V.; Bredas, J.L. Theoretical Study of the Local and Charge-Transfer Excitations in Model Complexes of Pentacene-C60 Using Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2014, 10, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Bredas, J.L.; Coropceanu, V. Description of the Charge Transfer States at the Pentacene/C60 Interface: Combining Range-Separated Hybrid Functionals with the Polarizable Continuum Model. J. Phys. Chem. Lett. 2016, 7, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yi, Y.P.; Zhang, C.R.; Aziz, S.G.; Coropceanu, V.; Bredas, J.L. Impact of Electron De localization on the Nature of the Charge-Transfer States in Model Pentacene/C-60 Interfaces: A Density Functional Theory Study. J. Phys. Chem. C 2014, 118, 27648–27656. [Google Scholar] [CrossRef]
- Muraoka, A.; Fujii, M.; Mishima, K.; Matsunaga, H.; Benten, H.; Ohkita, H.; Ito, S.; Yamashita, K. Investigations on the charge transfer mechanism at donor/acceptor interfaces in the quest for descriptors of organic solar cell performance. Phys. Chem. Chem. Phys. 2018, 20, 12193–12199. [Google Scholar] [CrossRef]
- Tsuneda, T.; Singh, R.K.; Nakata, A. Relationship between orbital energy gaps and excitation energies for long-chain systems. J. Comput. Chem. 2016, 37, 1451–1462. [Google Scholar] [CrossRef]
- Tsuneda, T.; Singh, R.K.; Nakata, A. On low-lying excited states of extended nanographenes. J. Comput. Chem. 2017, 38, 2020–2029. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, 1133. [Google Scholar] [CrossRef]
- Savin, A. On degeneracy, near-degeneracy and density functional theory. In Recent Developments and Applications of Modern Density Functional Theory; Seminario, J.M., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; p. 327. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Tsuneda, T.; Song, J.W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef]
- Chiba, M.; Tsuneda, T.; Hirao, K. Long-range corrected time-dependent density functional study on fluorescence of 4,4′-dimethylaminobenzonitrile. J. Chem. Phys. 2007, 126, 034504. [Google Scholar] [CrossRef] [PubMed]
- Chiba, M.; Tsuneda, T.; Hirao, K. Excited state geometry optimizations by analytical energy gradient of long-range corrected time-dependent density functional theory. J. Chem. Phys. 2006, 124, 144106. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Okano, Y.; Sato, T.; Kawashima, Y.; Tsuneda, T.; Hirao, K. Water cluster anions studied by the long-range corrected density functional theory. J. Phys. Chem. A 2008, 112, 9845–9853. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, M.; Tsuneda, T.; Hirao, K. A density functional study of van der Waals interactions. J. Chem. Phys. 2002, 117, 6010–6015. [Google Scholar] [CrossRef]
- Tokura, S.; Tsuneda, T.; Hirao, K. Long-range-corrected time-dependent density functional study on electronic spectra of five-membered ring compounds and free-base porphyrin. J. Theor. Comput. Chem. 2006, 5, 925–944. [Google Scholar] [CrossRef]
- Song, J.-W.; Hirosawa, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional calculations of chemical reactions: Redetermination of parameter. J. Chem. Phys. 2007, 126, 154105. [Google Scholar] [CrossRef]
- Sato, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional study on weakly bound systems: Balanced descriptions of various types of molecular interactions. J. Chem. Phys. 2007, 126, 234114. [Google Scholar] [CrossRef]
- Sato, T.; Tsuneda, T.; Hirao, K. Van der Waals interactions studied by density functional theory. Mol. Phys. 2005, 103, 1151–1164. [Google Scholar] [CrossRef]
- Sato, T.; Tsuneda, T.; Hirao, K. A density-functional study on pi-aromatic interaction: Benzene dimer and naphthalene dimer. J. Chem. Phys. 2005, 123, 104307. [Google Scholar] [CrossRef]
- Kamiya, M.; Sekino, H.; Tsuneda, T.; Hirao, K. Nonlinear optical property calculations by the long-range-corrected coupled-perturbed Kohn-Sham method. J. Chem. Phys. 2005, 122, 234111. [Google Scholar] [CrossRef]
- Song, J.-W.; Watson, M.A.; Sekino, H.; Hirao, K. The Effect of Silyl and Phenyl Functional Group End Caps on the Nonlinear Optical Properties of Polyynes: A Long-Range Corrected Density Functional Theory Study. Int. J. Quantum Chem. 2009, 109, 2012–2022. [Google Scholar] [CrossRef]
- Ahn, D.-H.; Song, J.-W. Assessment of long-range corrected density functional theory on the absorption and vibrationally resolved fluorescence spectrum of carbon nanobelts. J. Comput. Chem. 2021, 42, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zou, W.; Liu, W.; Xiao, Y.; Peng, D.; Song, B.; Liu, C. Time-dependent four-component relativistic density-functional theory for excitation energies. II. The exchange-correlation kernel. J. Chem. Phys. 2005, 123, 054102. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Ahn, M.; Bhattarai, K.; Wee, K.R.; Ahn, D.-H.; Song, J.-W. UV/Vis absorption spectrum calculations of benzo-1,2-dipyrene isomer using long-range corrected density functional theory. Chem. Phys. Lett. 2020, 761, 138023. [Google Scholar] [CrossRef]
- Kar, R.; Borpuzari, M.P.; Song, J.W.; Hirao, K. Molecules relevant for organic photovoltaics: A range-separated density functional study. Mol. Phys. 2015, 113, 2930–2938. [Google Scholar] [CrossRef]
- Kar, R.; Song, J.W.; Hirao, K. Long-range corrected functionals satisfy Koopmans’ theorem: Calculation of correlation and relaxation energies. J. Comput. Chem. 2013, 34, 958–964. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef]
- Dreuw, A.; Weisman, J.L.; Head-Gordon, M. Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange. J. Chem. Phys. 2003, 119, 2943–2946. [Google Scholar] [CrossRef]
- Tozer, D.J.; Handy, N.C. Improving virtual Kohn-Sham orbitals and eigenvalues: Application to excitation energies and static polarizabilities. J. Chem. Phys. 1998, 109, 10180–10189. [Google Scholar] [CrossRef]
- Song, J.-W.; Hirao, K. What makes differences between intra- and inter-molecular charge transfer excitations in conjugated long-chained polyene? EOM-CCSD and LC-BOP study. Theor. Chem. Acc. 2013, 133, 1438. [Google Scholar] [CrossRef]
- Song, J.-W.; Hirao, K. Is charge-transfer excitation through a polyalkane single-bond chain an intramolecular charge-transfer?: EOM-CCSD and LC-BOP study. Chem. Phys. Lett. 2022, 796, 139563. [Google Scholar] [CrossRef]
- Stein, T.; Eisenberg, H.; Kronik, L.; Baer, R. Fundamental Gaps in Finite Systems from Eigenvalues of a Generalized Kohn-Sham Method. Phys. Rev. Lett. 2010, 105, 266802. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef]
- Stein, T.; Kronik, L.; Baer, R. Prediction of charge-transfer excitations in coumarin-based dyes using a range-separated functional tuned from first principles. J. Chem. Phys. 2009, 131, 244119. [Google Scholar] [CrossRef]
- Oviedo, M.B.; Ilawe, N.V.; Wong, B.M. Polarizabilities of π-Conjugated Chains Revisited: Improved Results from Broken-Symmetry Range-Separated DFT and New CCSD(T) Benchmarks. J. Chem. Theory Comput. 2016, 12, 3593–3602. [Google Scholar] [CrossRef]
- Xu, L.; Kumar, A.; Wong, B.M. Linear polarizabilities and second hyperpolarizabilities of streptocyanines: Results from broken-Symmetry DFT and new CCSD(T) benchmarks. J. Comput. Chem. 2018, 39, 2350–2359. [Google Scholar] [CrossRef]
- Autschbach, J.; Srebro, M. Delocalization Error and “Functional Tuning” in Kohn–Sham Calculations of Molecular Properties. Acc. Chem. Res. 2014, 47, 2592–2602. [Google Scholar] [CrossRef]
- Körzdörfer, T.; Sears, J.S.; Sutton, C.; Brédas, J.-L. Long-range corrected hybrid functionals for π-conjugated systems: Dependence of the range-separation parameter on conjugation length. J. Chem. Phys. 2011, 135, 204107. [Google Scholar] [CrossRef]
- Refaely-Abramson, S.; Baer, R.; Kronik, L. Fundamental and excitation gaps in molecules of relevance for organic photovoltaics from an optimally tuned range-separated hybrid functional. Phys. Rev. B 2011, 84, 075144. [Google Scholar] [CrossRef]
- Sun, H.; Autschbach, J. Influence of the Delocalization Error and Applicability of Optimal Functional Tuning in Density Functional Calculations of Nonlinear Optical Properties of Organic Donor–Acceptor Chromophores. ChemPhysChem 2013, 14, 2450–2461. [Google Scholar] [CrossRef]
- de Queiroz, T.B.; Kümmel, S. Charge-transfer excitations in low-gap systems under the influence of solvation and conformational disorder: Exploring range-separation tuning. J. Chem. Phys. 2014, 141, 084303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zheng, X.; Li, C.; Yang, W. Orbital relaxation effects on Kohn–Sham frontier orbital energies in density functional theory. J. Chem. Phys. 2015, 142, 154113. [Google Scholar] [CrossRef] [PubMed]
- Casida, M.E. Recent Advances in Density Functional Methods; Part 1; Chong, D.P., Ed.; World Scientific: Singapore, 1995. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Wong, B.M. Optoelectronic Properties of Carbon Nanorings: Excitonic Effects from Time-Dependent Density Functional Theory. J. Phys. Chem. C Nanomater. Interfaces 2009, 113, 21921–21927. [Google Scholar] [CrossRef]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. [Google Scholar] [CrossRef]
- Perdew, J.P.; Parr, R.G.; Levy, M.; Balduz, J.L. Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy. Phys. Rev. Lett. 1982, 49, 1691–1694. [Google Scholar] [CrossRef]
- Perdew, J.P.; Levy, M. Physical Content of the Exact Kohn-Sham Orbital Energies: Band-Gaps and Derivative Discontinuities. Phys. Rev. Lett. 1983, 51, 1884–1887. [Google Scholar] [CrossRef]
- Sham, L.J.; Schluter, M. Density-Functional Theory of the Energy-Gap. Phys. Rev. Lett. 1983, 51, 1888–1891. [Google Scholar] [CrossRef]
- Casida, M.E. Correlated optimized effective-potential treatment of the derivative discontinuity and of the highest occupied Kohn-Sham eigenvalue: A Janak-type theorem for the optimized effective-potential model. Phys. Rev. B 1999, 59, 4694–4698. [Google Scholar] [CrossRef]
- Casida, M.E. Generalization of the Optimized-Effective-Potential Model to Include Electron Correlation—A Variational Derivation of the Sham-Schluter Equation for the Exact Exchange-Correlation Potential. Phys. Rev. A 1995, 51, 2005–2013. [Google Scholar] [CrossRef]
- Duffy, P.; Chong, D.P.; Casida, M.E.; Salahub, D.R. Assessment of Kohn-Sham Density-Functional Orbitals as Approximate Dyson Orbitals for the Calculation of Electron-Momentum-Spectroscopy Scattering Cross-Sections. Phys. Rev. A 1994, 50, 4707–4728. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Cohen, A.J.; Mori-Sanchez, P. Derivative discontinuity, bandgap and lowest unoccupied molecular orbital in density functional theory. J. Chem. Phys. 2012, 136, 204111. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Fractional charge perspective on the band gap in density-functional theory. Phys. Rev. B 2008, 77, 115123. [Google Scholar] [CrossRef]
- Sagvolden, E.; Perdew, J.P. Discontinuity of the exchange-correlation potential: Support for assumptions used to find it. Phys. Rev. A 2008, 77, 012517. [Google Scholar] [CrossRef]
- Perdew, J.P. What Do the Kohn-Sham Orbital Energies Mean? How Do Atoms Dissociate? In Density Functional Methods in Physics; Dreizler, R.M., Providencia, J., Eds.; Plenum: New York, NY, USA, 1985; Volume 123, p. 265. [Google Scholar]
- Sham, L.J.; Schluter, M. Density-Functional Theory of the Band-Gap. Phys. Rev. B 1985, 32, 3883–3889. [Google Scholar] [CrossRef]
- Janak, J.F. Proof That Delta-E-Delta-Ni=Epsilon-I in Density-Functional Theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. 3. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | -X(HX) | Intra | Inter | n | -X(HX) | Intra | Inter |
|---|---|---|---|---|---|---|---|
| 2 | -NH2(NH3) | 0.29 | 0.48 | 6 | -NH2(NH3) | 0.21 | 0.48 |
| -OH(H2O) | 0.30 | 0.48 | -OH(H2O) | 0.21 | 0.49 | ||
| -Cl(HCl) | 0.29 | 0.48 | -Cl(HCl) | 0.20 | 0.48 | ||
| -CHO(CH2O) | 0.29 | 0.46 | -CHO(CH2O) | 0.20 | 0.47 | ||
| -CN(HCN) | 0.29 | 0.48 | -CN(HCN) | 0.20 | 0.49 | ||
| -NO2(HNO2) | 0.30 | 0.47 | -NO2(HNO2) | 0.20 | 0.47 | ||
| 3 | -NH2(NH3) | 0.26 | 0.48 | 7 | -NH2(NH3) | 0.19 | 0.48 |
| -OH(H2O) | 0.27 | 0.49 | -OH(H2O) | 0.20 | 0.49 | ||
| -Cl(HCl) | 0.26 | 0.48 | -Cl(HCl) | 0.19 | 0.48 | ||
| -CHO(CH2O) | 0.26 | 0.47 | -CHO(CH2O) | 0.19 | 0.47 | ||
| -CN(HCN) | 0.26 | 0.47 | -CN(HCN) | 0.19 | 0.49 | ||
| -NO2(HNO2) | 0.26 | 0.47 | -NO2(HNO2) | 0.19 | 0.47 | ||
| 4 | -NH2(NH3) | 0.24 | 0.48 | 8 | -NH2(NH3) | 0.18 | 0.48 |
| -OH(H2O) | 0.24 | 0.49 | -OH(H2O) | 0.18 | 0.49 | ||
| -Cl(HCl) | 0.24 | 0.48 | -Cl(HCl) | 0.18 | 0.48 | ||
| -CHO(CH2O) | 0.24 | 0.47 | -CHO(CH2O) | 0.18 | 0.47 | ||
| -CN(HCN) | 0.24 | 0.49 | -CN(HCN) | 0.18 | 0.49 | ||
| -NO2(HNO2) | 0.24 | 0.47 | -NO2(HNO2) | 0.18 | 0.47 | ||
| 5 | -NH2(NH3) | 0.22 | 0.48 | ||||
| -OH(H2O) | 0.22 | 0.49 | |||||
| -Cl(HCl) | 0.22 | 0.48 | |||||
| -CHO(CH2O) | 0.22 | 0.47 | |||||
| -CN(HCN) | 0.22 | 0.49 | |||||
| -NO2(HNO2) | 0.22 | 0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, H.-S.; Ahn, D.-H.; Song, J.-W. Why Does the Optimal Tuning Method of the Range Separation Parameter of a Long-Range Corrected Density Functional Fail in Intramolecular Charge Transfer Excitation Calculations? Molecules 2024, 29, 4423. https://doi.org/10.3390/molecules29184423
Bae H-S, Ahn D-H, Song J-W. Why Does the Optimal Tuning Method of the Range Separation Parameter of a Long-Range Corrected Density Functional Fail in Intramolecular Charge Transfer Excitation Calculations? Molecules. 2024; 29(18):4423. https://doi.org/10.3390/molecules29184423
Chicago/Turabian StyleBae, Han-Seok, Dae-Hwan Ahn, and Jong-Won Song. 2024. "Why Does the Optimal Tuning Method of the Range Separation Parameter of a Long-Range Corrected Density Functional Fail in Intramolecular Charge Transfer Excitation Calculations?" Molecules 29, no. 18: 4423. https://doi.org/10.3390/molecules29184423