


Towards Nickel–NHC Fluoro Complexes—Synthesis of Imidazolium Fluorides and Their Reactions with Nickelocene



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of the Imidazolium Fluoride Salts 1a–1c
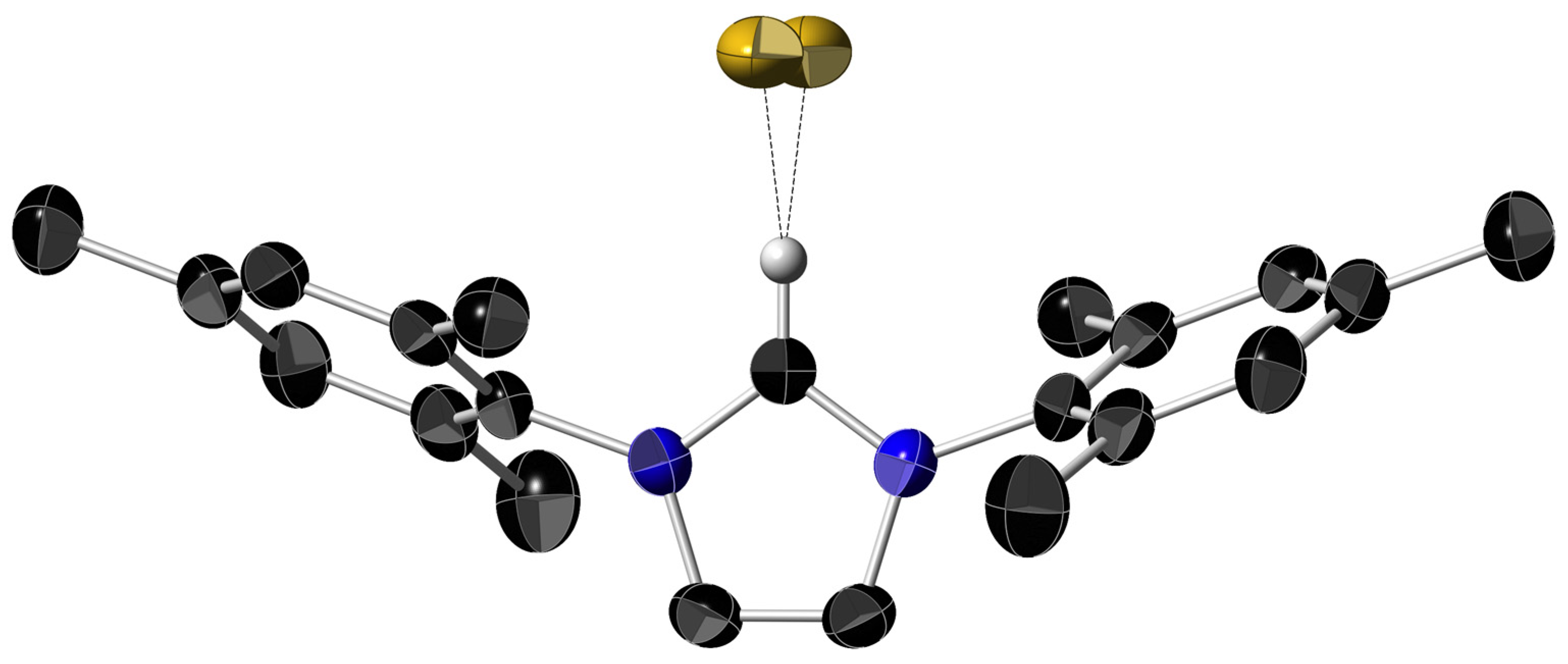
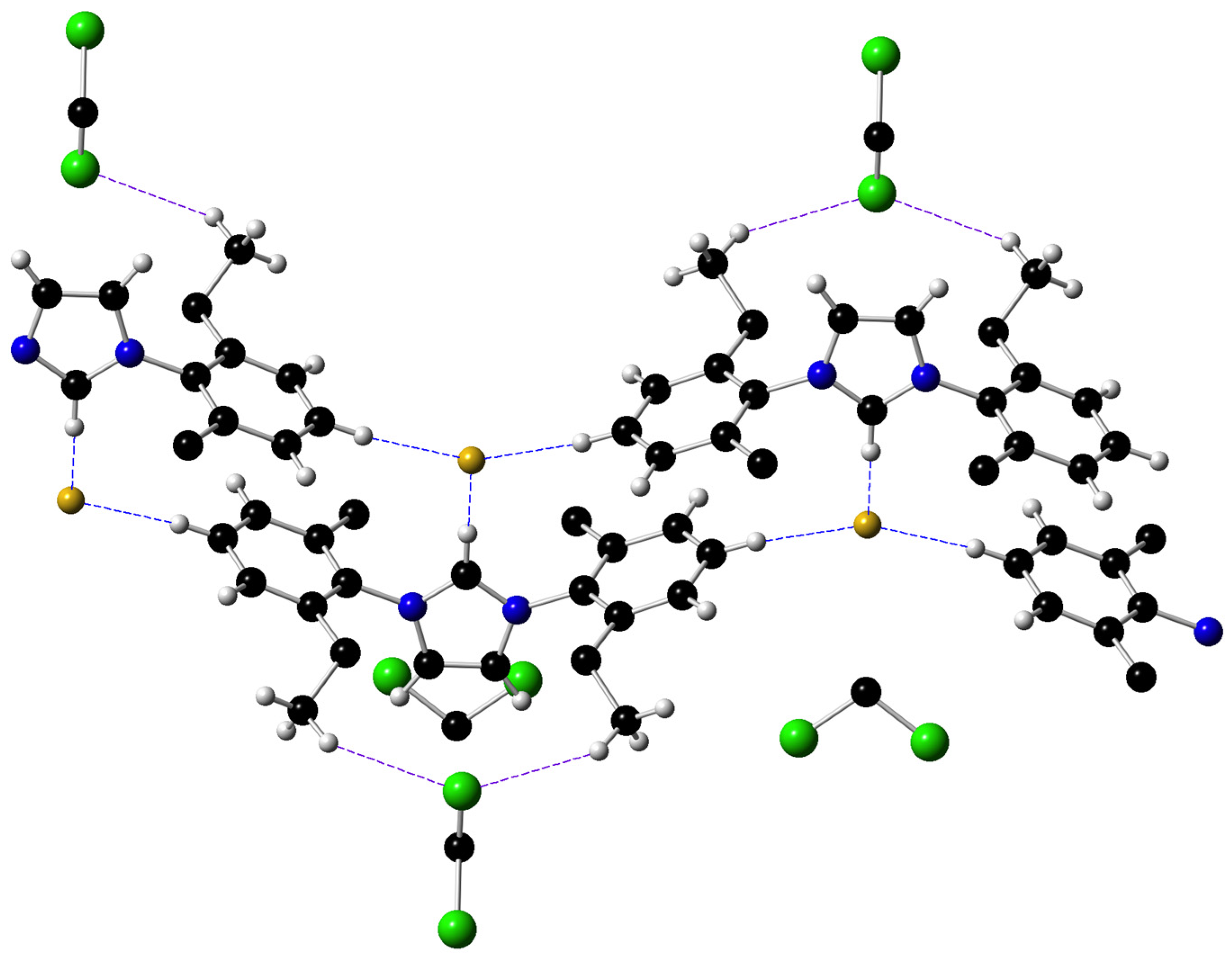
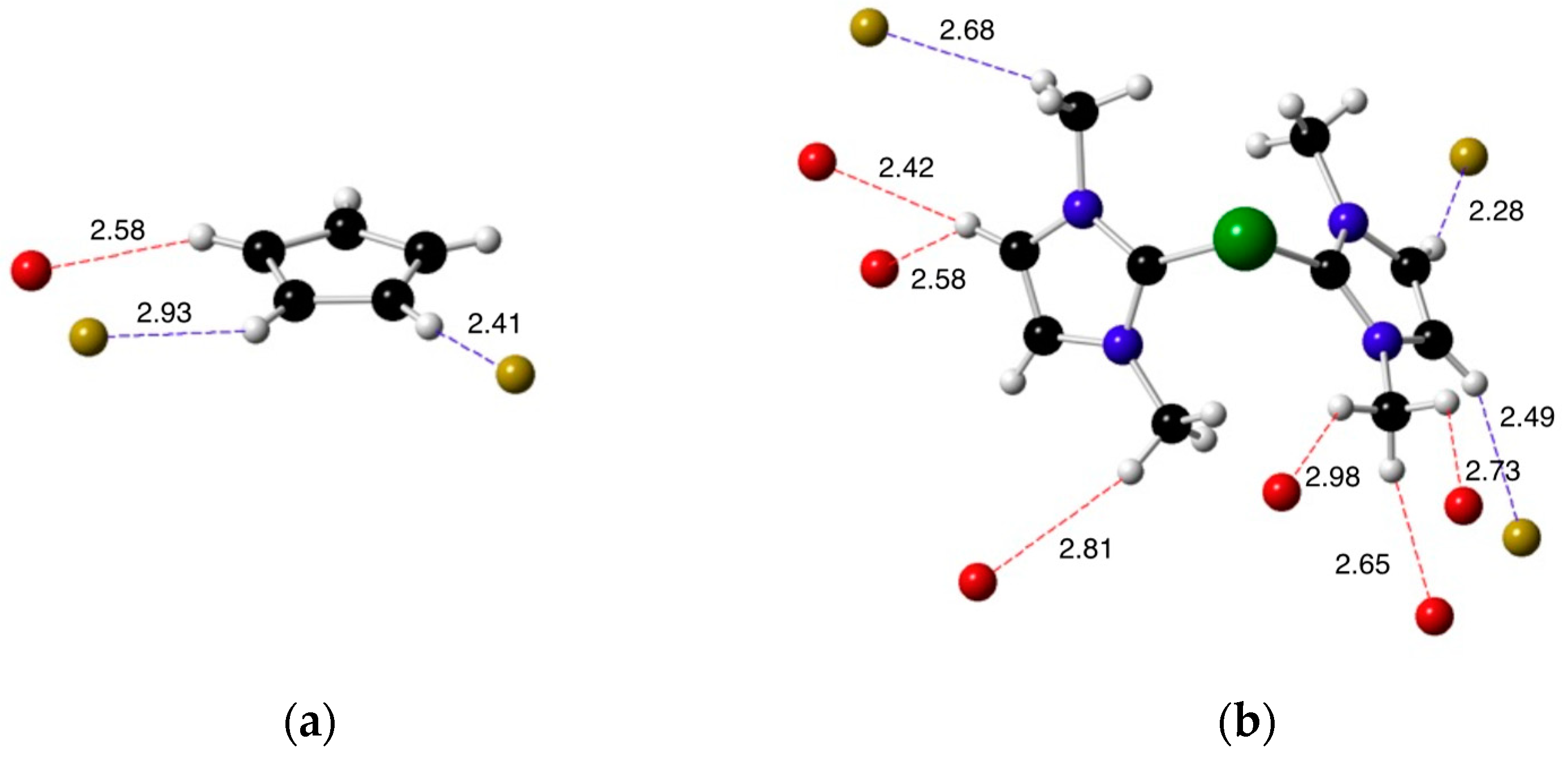
2.2. X-ray Diffraction Studies on the Imidazolium Fluoride Salts 1b and 1c
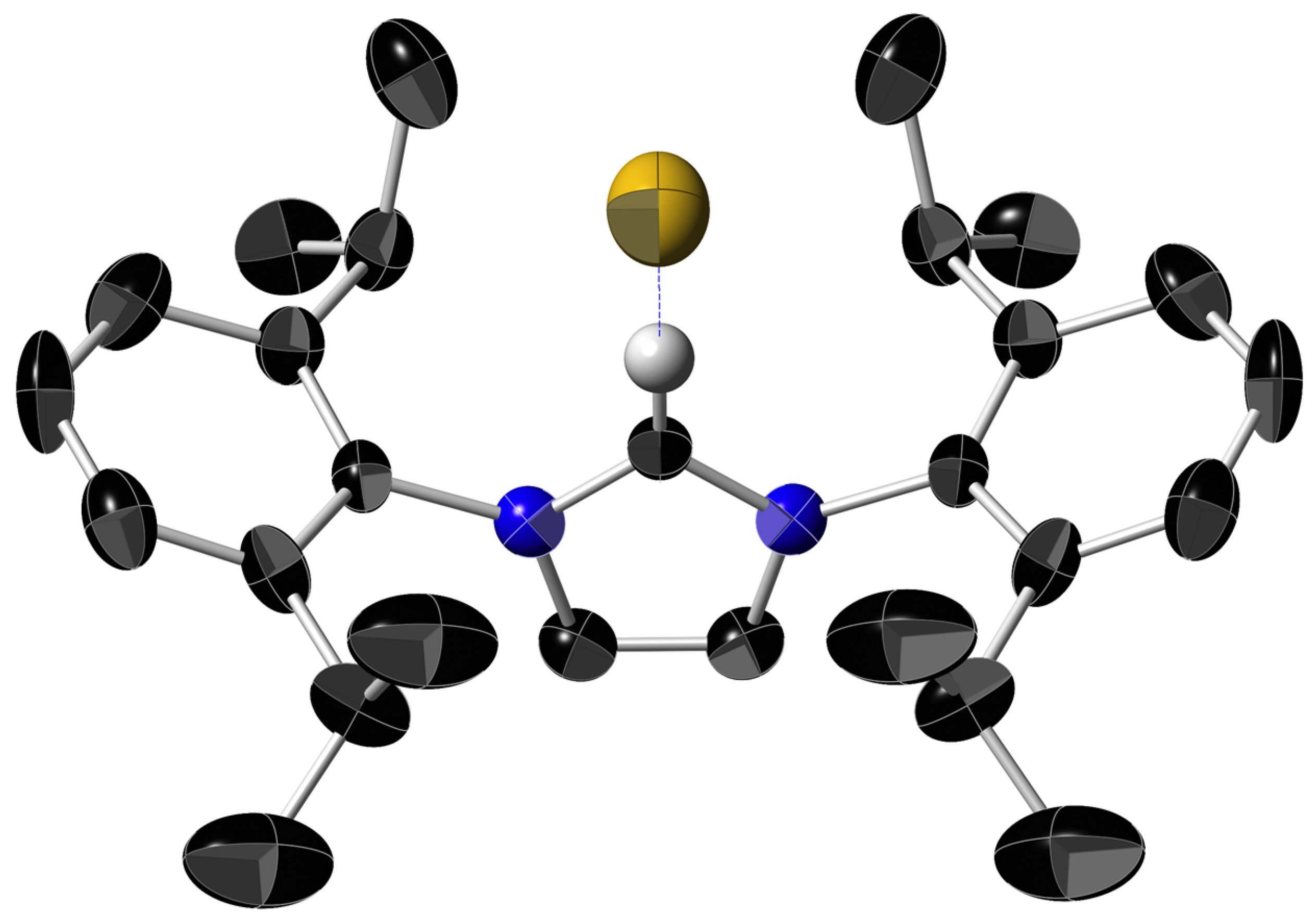
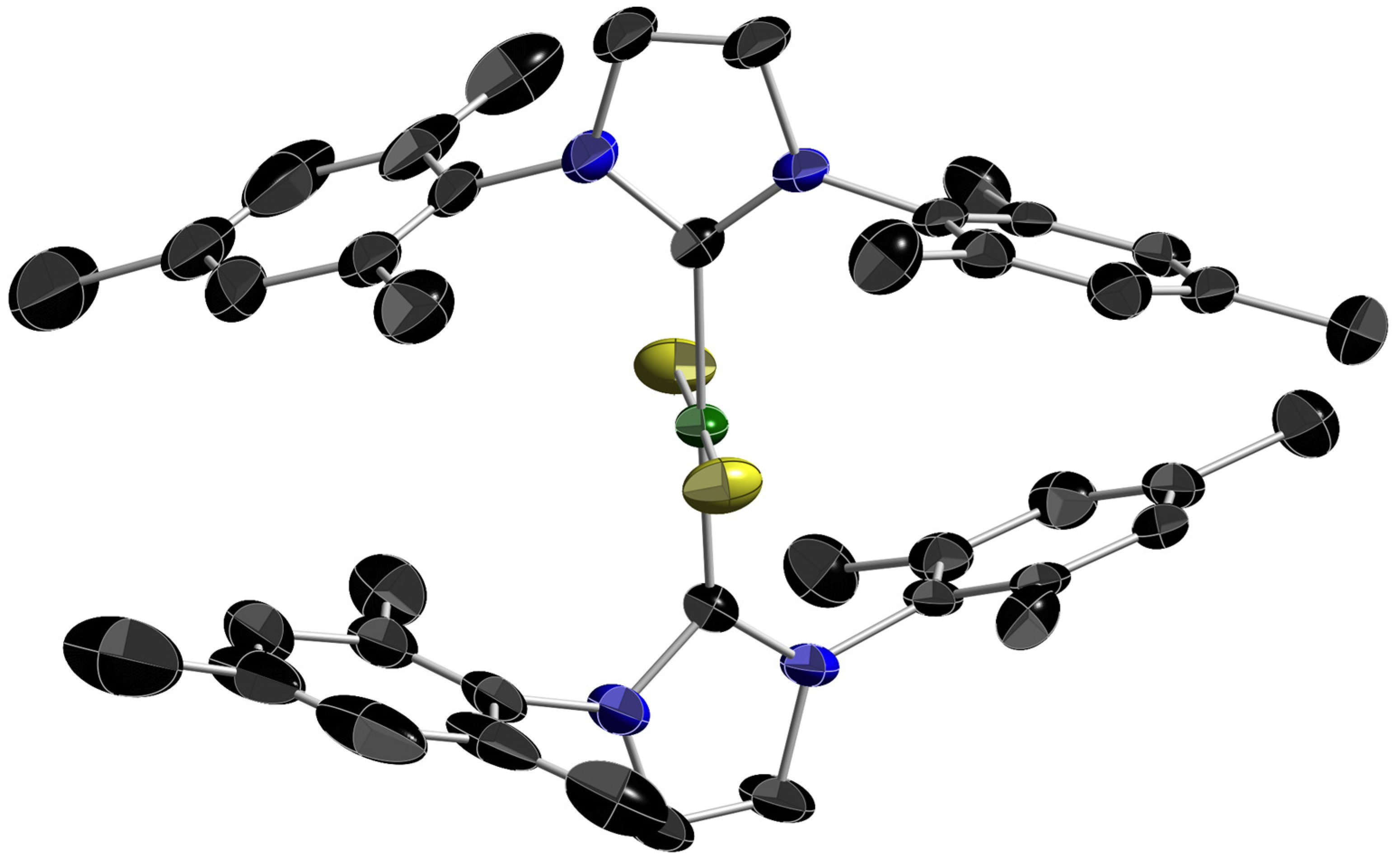
2.3. Reactions of Imidazolium Fluoride 1a with Nickelocene and an X-ray Diffraction Study of [Ni(Cp)(IMe)2]+ F−, 2
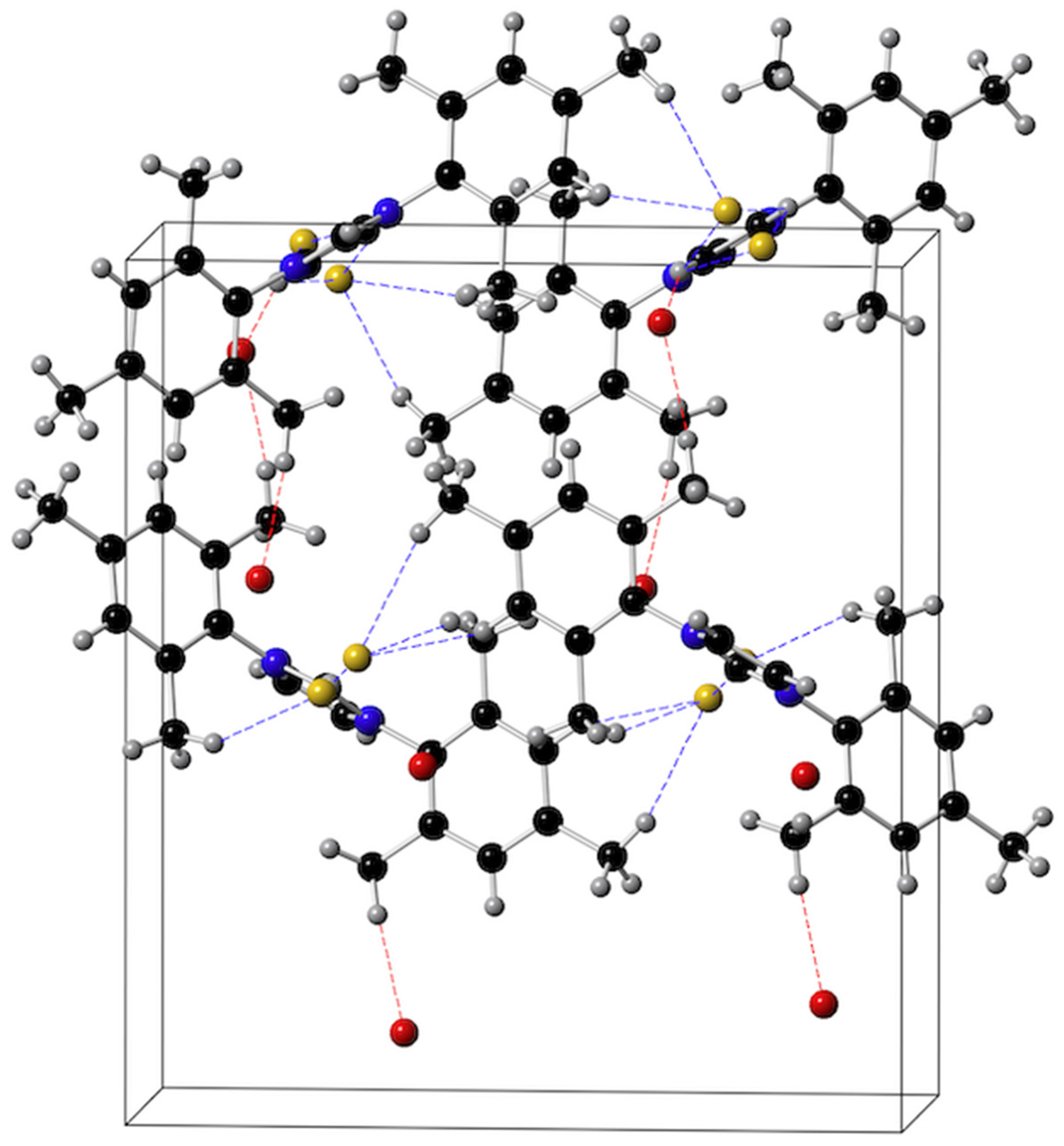
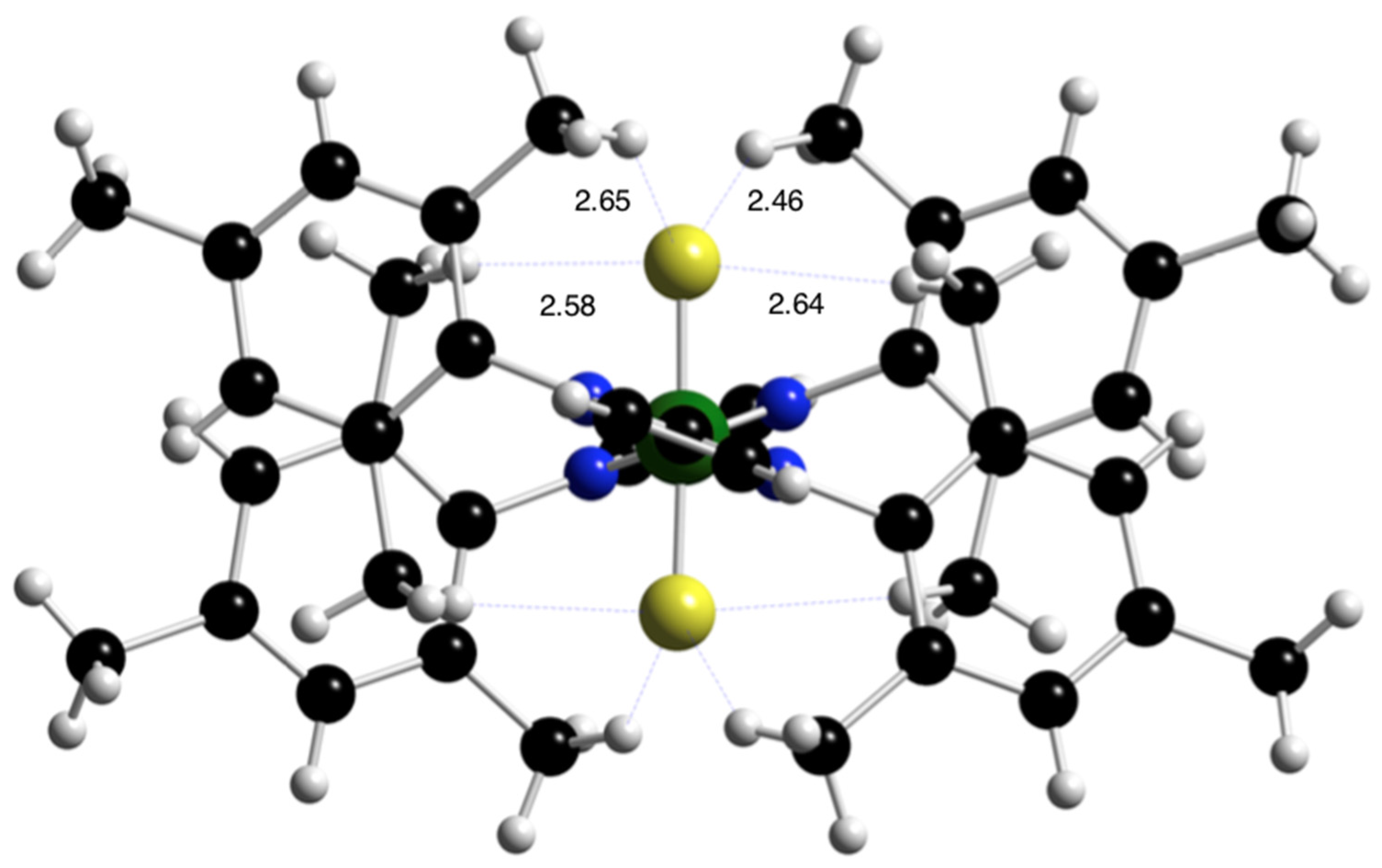
2.4. Reactions of the Imidazolium Fluorides 1b and 1c with Nickelocene and an X-ray Diffraction Study of trans-[NiF2(IMes)2], 3a
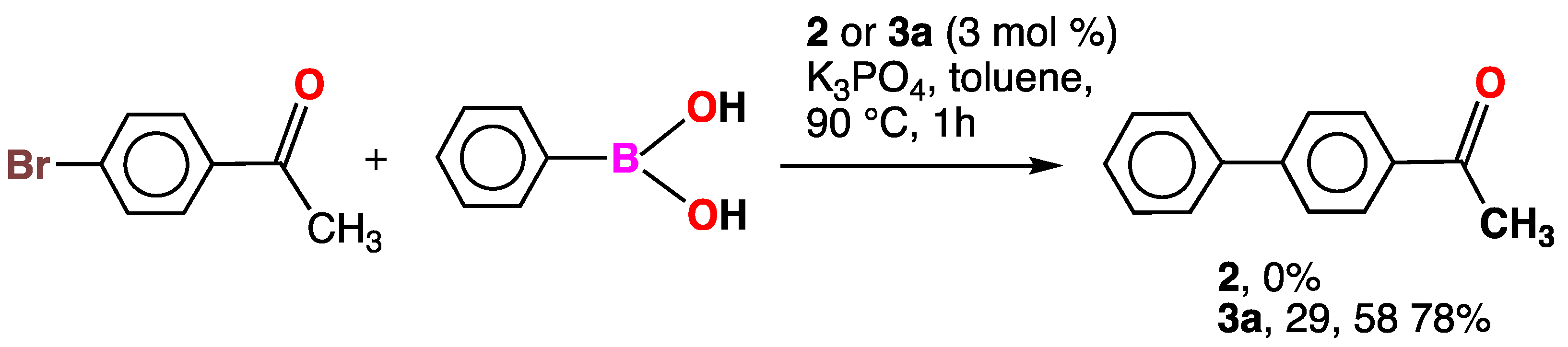
2.5. Catalytic Tests
3. Materials and Methods
3.1. Syntheses and Spectroscopic Data
3.2. N,N′-bis-(2,4,6-Trimethylphenyl)imidazolium Fluoride, [IMesH+ F−], 1b
3.3. N,N′-bis-(2,6-Diisopropylphenyl)imidazolium Fluoride, [IPrH+ F−], 1c
3.4. Synthesis of [Ni(Cp)(IMe)2]+ F−, 2
3.5. Synthesis of 3a, trans-[NiF2(IMes)2]
3.6. Catalytic Studies on the Reaction of 4-Bromoacetophenone with Phenylboronic Acid
3.7. Ligand Lability Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Dolbier, W.R., Jr. Fluorine chemistry at the millennium. J. Fluorine Chem. 2005, 126, 157–163. [Google Scholar] [CrossRef]
- Zhou, L. Recent Advances in C–F Bond Cleavage Enabled by Visible Ligh Photoredox Catalysis. Molecules 2021, 26, 7051. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.M.; Pedersen, C.M. Vessel effects in organic chemical reactions; a century old, overlooked phenomenon. Chem. Sci. 2022, 13, 6181–6196. [Google Scholar] [CrossRef] [PubMed]
- Grushkin, V.V. The Organometallic Fluorine Chemistry of Palladium and Rhodium: Studies toward Aromatic Fluorination. Acc. Chem. Res. 2010, 43, 160–171. [Google Scholar] [CrossRef]
- Murphy, E.F.; Ramaswarmy, M.; Roesky, H.W. Organometallic Fluorides: Compounds Containing Carbon-Metal–Fluorine Fragments of d-Block Metals. Chem. Rev. 1997, 97, 3425–3468. [Google Scholar] [CrossRef]
- Roesky, H.W. Playing the Keyboard of Fluorine Chemistry. Inorg. Chem. 1999, 38, 5934–5943. [Google Scholar] [CrossRef]
- Jasim, N.A.; Perutz, R.N.; Whitwood, A.C.; Braun, T.; Izundu, J.; Neumann, B.; Rothfeld, S.; Stammler, H.-G. Complexes Contrasting Reactivity of Fluoropyridines at Palladium and Platinum: C−F Oxidative Addition at Palladium, P−C and C−F Activation at Platinum. Organometallics 2004, 26, 6140–6149. [Google Scholar] [CrossRef]
- Schaub, T.; Fischer, P.; Steffen, A.; Braun, T.; Radius, U.; Mix, A. C-F Activation of Fluorinated Arenes using NHC-Stabilized Nickel(0) Complexes: Selectivity and Mechanistic Investigations. J. Am. Chem. Soc. 2008, 130, 9304–9317. [Google Scholar] [CrossRef]
- Schaub, T.; Backes, M.; Radius, U. Catalytic C−C Bond Formation Accomplished by Selective C−F Activation of Perfluorinated Arenes. J. Am. Chem. Soc. 2006, 128, 15964–15965. [Google Scholar] [CrossRef]
- Rachor, S.G.; Müller, R.; Kaupp, M.; Braun, T. Hydrogen and Halogen Bonding to Au(I) Fluorido Complexes. Eur. J. Inorg. Chem. 2023, 26, e202200668. [Google Scholar] [CrossRef]
- Gauthier, R.; Nikolaos, V.; Tzouras, N.V.; Zhang, Z.; Bédard, S.; Saab, M.; Falivene, L.; Van Hecke, K.; Luigi Cavallo, L.; Nolan, S.P.; et al. Gold N-Heterocyclic Carbene Catalysts for the Hydrofluorination of Alkynes Using Hydrofluoric Acid: Reaction Scope, Mechanistic Studies and the Tracking of Elusive Intermediates. Chem. Eur. J. 2022, 22, e202103886. [Google Scholar] [CrossRef] [PubMed]
- Ritleng, V.; Barth, C.; Brenner, E.; Milosevic, S.; Chetcuti, M.J. Synthesis, structure, and solution dynamics of pentamethyl-cyclopentadienyl nickel complexes bearing N-heterocyclic carbene ligands. Organometallics 2008, 27, 4223–4228. [Google Scholar] [CrossRef]
- Ulm, F.; Poblador-Bahamonde, A.I.; Choppin, S.; Bellemin-Laponnaz, S.; Chetcuti, M.J.; Achard, T.; Ritleng, V. Synthesis, characterization, and catalytic application in aldehyde hydrosilylation of half- sandwich nickel complexes bearing (κ1-C)- and hemilabile (κ2-C,S)-thioether functionalized NHC ligands. Dalton Trans. 2018, 47, 17134–17145. [Google Scholar] [CrossRef] [PubMed]
- Abernethy, C.D.; Clyburne, J.A.C.; Cowley, A.H.; Jones, R.A. Reactions of Transition-Metal Metallocenes with Stable Carbenes. J. Am. Chem. Soc. 1999, 121, 2329–2330. [Google Scholar] [CrossRef]
- Ritleng, V.; Oertel, A.M.; Chetcuti, M.J. Half-sandwich NHC-nickel(II) complexes as pre-catalysts for the fast Suzuki coupling of aryl halides: A comparative study. Dalton Trans. 2010, 39, 8153–8160. [Google Scholar] [CrossRef]
- Oertel, A.M.; Ritleng, V.; Chetcuti, M.J. Synthesis and Catalytic Activity in Suzuki Coupling of Nickel Complexes Bearing n-Butyl- and Triethoxysilylpropyl-Substituted NHC Ligands: Toward the Heterogenization of Molecular Catalysts. Organometallics 2012, 7, 2829–2840. [Google Scholar] [CrossRef]
- Aloui, L.; Abidi, R.; Chetcuti, M.J. Synthesis and characterization of nickel-N-heterocyclic carbenes linked to a calix[6]arene platform and their applications in Suzuki-Miyaura cross-coupling catalysis. Inorg. Chim. Acta 2020, 505, 119494. [Google Scholar] [CrossRef]
- Zell, T.; Fischer, P.; Schmidt, D.; Radius, U. C–Br Activation of Aryl Bromides at Ni0(NHC)2: Stoichiometric Reactions, Catalytic Application in Suzuki–Miyaura Cross-Coupling, and Catalyst Degradation. Organometallics 2012, 31, 5065–5073. [Google Scholar] [CrossRef]
- Gu, S.; Du, J.; Huang, J.; Guo, Y.; Yang, L.; Xu, W.; Chen, W. Unsymmetrical NCN-pincer mononuclear and dinuclear nickel(II) complexes of N-heterocyclic carbene (NHC): Synthesis, structure and catalysis for Suzuki–Miyaura cross-coupling. Dalton Trans. 2017, 46, 586–594. [Google Scholar] [CrossRef]
- Rahman, M.M.; Zhao, Q.; Meng, G.; Szostak, R.; Szostak, M. [Ni(Np#)(η5-Cp)Cl]: Flexible, Sterically Bulky, Well-Defined, Highly Reactive Complex for Nickel-Catalyzed Cross-Coupling. Organometallics 2022, 41, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-Y.; Chan, K.-T.; Chang, Y.-C.; Chen, C.-Y.; Tu, C.-Y.; Hu, C.-H.; Lee, H.M. Unexpected Solvent-Induced Cis/Trans Isomerization and Catalytic Application of a Bis-bidentate Nickel(II) Complex with N-Heterocyclic Carbene and Amido Functionalities. Organometallics 2007, 26, 5826–5833. [Google Scholar] [CrossRef]
- Henrion, M.; Ritleng, V.; Chetcuti, M.J. From acetone metalation to the catalytic α-arylation of acyclic ketones with NHC–nickel(II) complexes. Chem. Commun. 2014, 50, 4624–4627. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Ruan, L.-X.; Rahman, A.; Shi, S.-L. Fast Enantio- and Chemoselective Arylation of Ketones with Organoboronic Esters Enabled by Nickel/N-Heterocyclic Carbene Catalysis. Ang. Chem. Int. Ed. 2021, 60, 5262–5267. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salas, J.A.; Marelli, E.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P. General and Mild Ni0-Catalyzed α-Arylation of Ketones Using Aryl Chlorides. Chem. Eur. J. 2015, 21, 3906–3909. [Google Scholar] [CrossRef]
- Ulm, F.; Shahane, S.; Truong-Phuoc, L.; Romero, T.; Papaefthimiou, V.; Chessé, M.; Chetcuti, M.J.; Pham-Huu, C.; Michon, C.; Ritleng, V. Half-Sandwich Nickel(II) NHC-Picolyl Complexes as Catalysts for the Hydrosilylation of Carbonyl Compounds: Evidence for NHC-Nickel Nanoparticles under Harsh Reaction Conditions. Eur. J. Inorg. Chem. 2021, 30, 3074–3082. [Google Scholar] [CrossRef]
- Chang, A.S.; Kawamura, K.E.; Henness, H.S.; Salpino, V.M.; Greene, J.C.; Zakharov, L.N.; Cook, A.K. (NHC)Ni(0)-Catalyzed Branched-Selective Alkene Hydrosilylation with Secondary and Tertiary Silanes. ACS Catal. 2022, 12, 11002–11014. [Google Scholar] [CrossRef]
- Junquera, L.B.; Puerta, M.C.; Valerga, P. R-Allyl Nickel(II) Complexes with Chelating N-Heterocyclic Carbenes: Synthesis, Structural Characterization, and Catalytic Activity. Organometallics 2012, 31, 2175–2183. [Google Scholar] [CrossRef]
- Ulm, F.; Djukic, J.-P.; Chetcuti, M.J.; Ritleng, V. Hydroboration of alkenes catalyzed by a nickel N-heterocyclic carbene complex: Reaction and mechanistic aspects. Chem. Eur. J. 2020, 26, 8916–8925. [Google Scholar] [CrossRef]
- Afandiyeva, M.; Kadam, A.A.; Wu, X.; Brennessel, W.W.; Kennedy, C.R. Synthesis, Structure, and Hydroboration Reactivity of Anionic Nickel(0) Complexes Supported by Bidentate NHC-Pyridone Ligands. Organometallics 2022, 41, 3014–3023. [Google Scholar] [CrossRef]
- Touney, E.E.; Van Hoveln, R.; Buttke, C.T.; Freidberg, M.D.; Guzei, I.A.; Schomaker, J.M. Heteroleptic Nickel Complexes for the Markovnikov-Selective Hydroboration of Styrenes. Organometallics 2016, 35, 3436–3439. [Google Scholar] [CrossRef]
- Lee, B.C.; Liu, C.-F.; Lin, L.Q.H.; Yap, K.Z.; Song, N.; Ko, C.H.M.; Chan, P.H.; Koh, M.J. N-Heterocyclic carbenes as privileged ligands for nickel-catalysed alkene functionalisation. Chem. Soc. Rev. 2023, 52, 2946–2991. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, T.; Hashimoto, S.; Ishizuka, K.; Nakamura, M. Highly Selective Biaryl Cross-Coupling Reactions between Aryl Halides and Aryl Grignard Reagents: A New Catalyst Combination of N-Heterocyclic Carbenes and Iron, Cobalt, and Nickel Fluorides. J. Am. Chem. Soc. 2009, 131, 11949–11963. [Google Scholar] [CrossRef] [PubMed]
- Schaub, T.; Backes, M.; Radius, U. Square-Planar (Pentafluorophenyl)nickel(II) Complexes by Derivatization of a C–F Activation Product. Eur. J. Inorg. Chem. 2008, 17, 2680–2690. [Google Scholar] [CrossRef]
- Jahnke, M.C.; Hahn, F.E. Chemistry of N-heterocyclic Carbene Ligands. Top. Organomet. Chem. 2010, 30, 95–129. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Krafczyk, R.; Schmutzler, R.; Craig, H.A.; Goerlich, J.R.; Marshall, W.J.; Unverzagt, M. Imidazolylidenes, imidazolinylidenes and imidazolidines. Tetrahedron 1999, 55, 14523–14534. [Google Scholar] [CrossRef]
- Hintermann, L. Expedient syntheses of the N-heterocyclic carbene precursor imidazolium salts Ipr·HCl, IMes·HCl and IXy·HCl. Belstein J. Org. Chem. 2007, 3, 22. [Google Scholar] [CrossRef]
- Oertel, A.M.; Ritleng, V.; Chetcuti, M.J. N′-Activation of N-Arylimidazoles: Facile Syntheses of N-Alkyl-N′-arylimidazolium Iodides from Less Expensive Chloro Substrates. Synthesis 2009, 10, 1647–1650. [Google Scholar] [CrossRef]
- Zhu, Z.-Q.; Jiang, M.-Y.; Zheng, C.-G.; Xiao, J.-C. Efficient synthesis of 1-alkyl-3-methylimidazolium fluorides and possibility of the existence of hydrogen bonding between fluoride anion and C(sp3)–H. J. Fluorine Chem. 2012, 133, 160–162. [Google Scholar] [CrossRef]
- Pilon, M.C.; Grushin, V. Synthesis and Characterization of Organopalladium Complexes Containing a Fluoro Ligand. Organometallics 1998, 17, 1774–1781. [Google Scholar] [CrossRef]
- Bouvet, S.; Pégot, B.; Marrot, J.; Magnier, E. Solvent free nucleophilic introduction of fluorine with [bmim][F]. Tetrahedron Lett. 2014, 55, 826–829. [Google Scholar] [CrossRef]
- Bouvet, S.; Pégot, B.; Diter, P.; Marrot, J.; Magnier, E. Oxidative desulfurization–fluorination reaction promoted by [bdmim][F] for the synthesis of difluorinated methyl ethers. Tetrahedron Lett. 2015, 56, 1682–1686. [Google Scholar] [CrossRef]
- Alič, B.; Petrovčic, J.; Jelen, J.; Tavčar, G.; Iskra, J. Renewable Reagent for Nucleophilic Fluorination. J. Org. Chem. 2022, 87, 5987–5993. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Wang, W.; Ritter, T. Deoxyfluorination of Phenols. J. Am. Chem. Soc. 2011, 133, 11482–11484. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, N.W.; Shen, X.; Li, J.; Ritter, T. AlkylFluor: Deoxyfluorination of Alcohols. Org. Lett. 2016, 18, 6102–6104. [Google Scholar] [CrossRef]
- Alič, B.; Tavčar, G. Reaction of N-heterocyclic carbene (NHC) with different HF sources and ratios—A free fluoride reagent based on imidazolium fluoride. J. Fluorine Chem. 2016, 192, 141–146. [Google Scholar] [CrossRef]
- Huang, H.; Zhong, J.; Tan, X.; Guo, X.; Yuan, B.; Lin, Y.; Francisco, J.S.; Zeng, X.G. New Insights into the Stability of Anhydrous 2H-Imidazolium Fluoride and its High Dissolution Capability toward a Strongly Hydrogen-Bonded Compound. J. Am. Chem. Soc. 2020, 142, 10314–10318. [Google Scholar] [CrossRef]
- Alič, B.; Tramšek, M.; Kokalj, A.; Tavcar, G. Discrete GeF5− Anion Structurally Characterized with a Readily Synthesized Imidazolium Based Naked Fluoride Reagent. Inorg. Chem. 2017, 56, 10070–10077. [Google Scholar] [CrossRef]
- Libri, S.; Jasim, N.A.; Perutzm, R.N.; Brammer, L. Metal Fluorides Form Strong Hydrogen Bonds and Halogen Bonds: Measuring Interaction Enthalpies and Entropies in Solution. J. Am. Chem. Soc. 2008, 130, 7842–7844. [Google Scholar] [CrossRef]
- Kelly, R.A.; Scott, N.M.; Díez-González, S.; Stevens, E.D.; Nolan, S.P. Simple Synthesis of CpNi(NHC)Cl Complexes (Cp = Cyclopentadienyl; NHC = N-Heterocyclic Carbene). Organometallics 2005, 24, 3442–3447. [Google Scholar] [CrossRef]
- Abernethy, C.D.; Cowley, A.H.; Jones, R.A. Reactions of Nickelocene with 1,3-dimesitylimidazolium chloride. J. Organomet. Chem. 2000, 596, 3–5. [Google Scholar] [CrossRef]
- Malan, F.P.; Singleton, E.; van Rooyen, P.H.; Conradie, J.; Landman, M. Synthesis, structure and DFT conformation analysis of CpNiX(NHC) and NiX2(NHC)2 (X = SPh or Br) complexes. J. Mol. Struct. 2017, 1147, 235–243. [Google Scholar] [CrossRef]
- Banach, L.; Gunka, P.A.; Zachara, J.; Buchowicz, W. Half-sandwich Ni(II) complexes [Ni(Cp)(X)(NHC)]: From an underestimated discovery to a new chapter in organonickel chemistry. Coord. Chem. Rev. 2019, 389, 19–58. [Google Scholar] [CrossRef]
- Bielinski, E.A.; Dai, W.; Guard, L.M.; Hazari, N.; Takase, M.K. Synthesis, properties, and reactivity of palladium and nickel NHC complexes supported by combinations of allyl, cyclopentadienyl, and indenyl ligands. Organometallics 2013, 32, 4025–4037. [Google Scholar] [CrossRef]
- Pankov, R.O.; Tarabrin, I.R.; Son, A.G.; Minyaev, M.E.; Prima, D.O.; Ananikov, V.P. Synthesis and comparative study of (NHCF)PdCl2Py and (NHCF)Ni(Cp)Cl complexes: Investigation of the electronic properties of NHC ligands and complex characteristics. Dalton Trans. 2024, 53, 12503–12518. [Google Scholar] [CrossRef]
- Oertel, A.M.; Ritleng, V.; Burr, L.; Chetcuti, M.J. Synthesis and Structural Characterization of Half-Sandwich Nickel Complexes Bearing Two Different N-Heterocyclic Carbene Ligands. Organometallics 2011, 30, 6685–6691. [Google Scholar] [CrossRef]
- Henrion, M.; Oertel, A.M.; Ritleng, V.; Chetcuti, M.J. Facile displacement of η5-cyclopentadienyl ligands from half-sandwich alkyl,NHC–nickel complexes: An original route to robust cis-C,C-nickel square planar complexes. Chem. Commun. 2013, 49, 6424–6426. [Google Scholar] [CrossRef]
- Oertel, A.M.; Ritleng, V.; Chetcuti, M.J.; Veiros, L.F. C-H Activation of Acetonitrile at Nickel: Ligand Flip and Conversion of N-Bound Acetonitrile into a C-Bound Cyanomethyl Ligand. J. Am. Chem. Soc. 2010, 132, 13588–13589. [Google Scholar] [CrossRef]
- Gibson, J.A.; Ibbott, D.G.; Janzen, A.F. Fluorine NMR spectra of sulfur, phosphorus, and silicon fluorides. Hydrolysis and intra-intermolecular mechanism of fluorine exchange. Can. J. Chem. 1973, 51, 3203–3210. [Google Scholar] [CrossRef]
- Klanberg, F.; Muetterties, E.L. Nuclear Magnetic Resonance Studies on Pentacoordinate Silicon Fluorides. Inorg. Chem. 1968, 7, 155–160. [Google Scholar] [CrossRef]
- Rosenau, C.P.; Jelier, B.J.; Gossert, A.D.; Togni, A. Exposing the Origins of Irreproducibility in Fluorine NMR Spectroscopy. Ang. Chem. 2018, 57, 9528–9533. [Google Scholar] [CrossRef]
- Finney, W.F.; Wilson, E.; Callender, A.; Morris, M.D.; Beck, L.W. Reexamination of Hexafluorosilicate Hydrolysis by 19F NMR and pH Measurement. Environ. Sci. Technol. 2006, 40, 2572–2577. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, S. Complex fluoroanions in solution. VII. Replacement of fluoride by trifluoroacetate. Can. J. Chem. 1978, 56, 343–347. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, S.; Li, Z.; Wang, Q.; Weng, L. Direct synthesis of cis-dihalido-bis(NHC) complex of nickel(II) and catalytic application in olefin addition polymerization: Effect of halogen co-ligands and density functional theory study. Dalton Trans. 2013, 42, 12020–12030. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Xu, Y.-C.; Xie, L.-Z.; Sun, H.-M.; Shen, Q.; Zhang, Y. Controlled synthesis of nickel(II) dihalides bearing two different or identical N-heterocyclic carbene ligands and the influence of carbene ligands on their structures and catalysis. Dalton Trans. 2011, 40, 4697. [Google Scholar] [CrossRef]
- Ritleng, V.; Brenner, E.; Chetcuti, M.J. Preparation of a N-heterocyclic carbene nickel(II) complex. Synthetic experiments in current organic and organometallic chemistry. J. Chem. Educ. 2008, 85, 1646–1648. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97 Program for Crystal Structure Determination. Acta Crystallogr. 1990, 46, 467–473. [Google Scholar] [CrossRef]
- M86-E01078 APEX2 User Manual; Bruker AXS Inc.: Madison, WI, USA, 2006.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (Å or °) | 1a, [(IMe)+ F−] a | 1b, [(IMesH)+ F−] | 1c, [(IPrH)+ F−] |
|---|---|---|---|
| C–Himid … F | 1.92 and 2.04 | 1.85 | 1.84 |
| C=Cimid | 1.331 (3) | 1.341 (6) | 1.360 (17) |
| C(1)imid–N | 1.318 (3) | 1.332 (3) | 1.328 (9) |
| C(2)imid–N | 1.362 (3) | 1.377 (3) | 1.355 (11) |
| b Ar–imid. least sq. planes angle | – | 70.2 | 82.0 |
| Parameter (Å, Bond-Distances; Angles,°) | 2, [Ni(Cp)(IMe)2+] F−. |
|---|---|
| a Ni–Ccarb | 1.892 (6) |
| a C=Cimid | 1.320 (11) |
| a C(1)imid–N | 1.350 (8) |
| a C(2)imid–N and C(2′)imid–N | 1.388 (9) |
| a N–CMe | 1.463 (9) |
| a Ni–Cp | 2.124 (6) |
| Ni–Cpcentroid | 1.752 |
| Ccarb–Ni–Ccarb | 94.5 (2) |
| Ccarb–Ni–Cpcentroid | 132.3, 133.1 |
| b IMe(1)–IMe(2) angle | 94.5 |
| Parameter | 3a | A | B | C |
|---|---|---|---|---|
| Ni–Ccarb | 1.905 (4) | 1.932 (8), 1.911 (8) | 1.924 (2), 1.933 (2) | 1.911 (6), 1.905 6) |
| Ni–F | 1.816 (2) | 1.856 (4) | 1.891 (1) | 1.856 (3) |
| F–Ni–F | 178.3 (18) | 178.6 (3) | 175.5 (1) 1 | 177.7 (2) 1 |
| F–Ni–Ccarb | 89.23 (13), 90.79 (13) | 89.1 (2), 88.0 (3) | 90.2 (1), 88.6 (1) | 89.4 (2), 89.0 (2) |
| Ccarb–Ni–Ccarb | 178.3 (18) | 175.0 (3) | - | 175.0 (2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wills, S.S.; Bailly, C.; Chetcuti, M.J. Towards Nickel–NHC Fluoro Complexes—Synthesis of Imidazolium Fluorides and Their Reactions with Nickelocene. Molecules 2024, 29, 4493. https://doi.org/10.3390/molecules29184493
Wills SS, Bailly C, Chetcuti MJ. Towards Nickel–NHC Fluoro Complexes—Synthesis of Imidazolium Fluorides and Their Reactions with Nickelocene. Molecules. 2024; 29(18):4493. https://doi.org/10.3390/molecules29184493
Chicago/Turabian StyleWills, Siobhan S., Corinne Bailly, and Michael J. Chetcuti. 2024. "Towards Nickel–NHC Fluoro Complexes—Synthesis of Imidazolium Fluorides and Their Reactions with Nickelocene" Molecules 29, no. 18: 4493. https://doi.org/10.3390/molecules29184493