
Development and Evaluation of a Cost-Effective, Carbon-Based, Extended-Release Febuxostat Tablet

 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Method of Analysis by HPLC
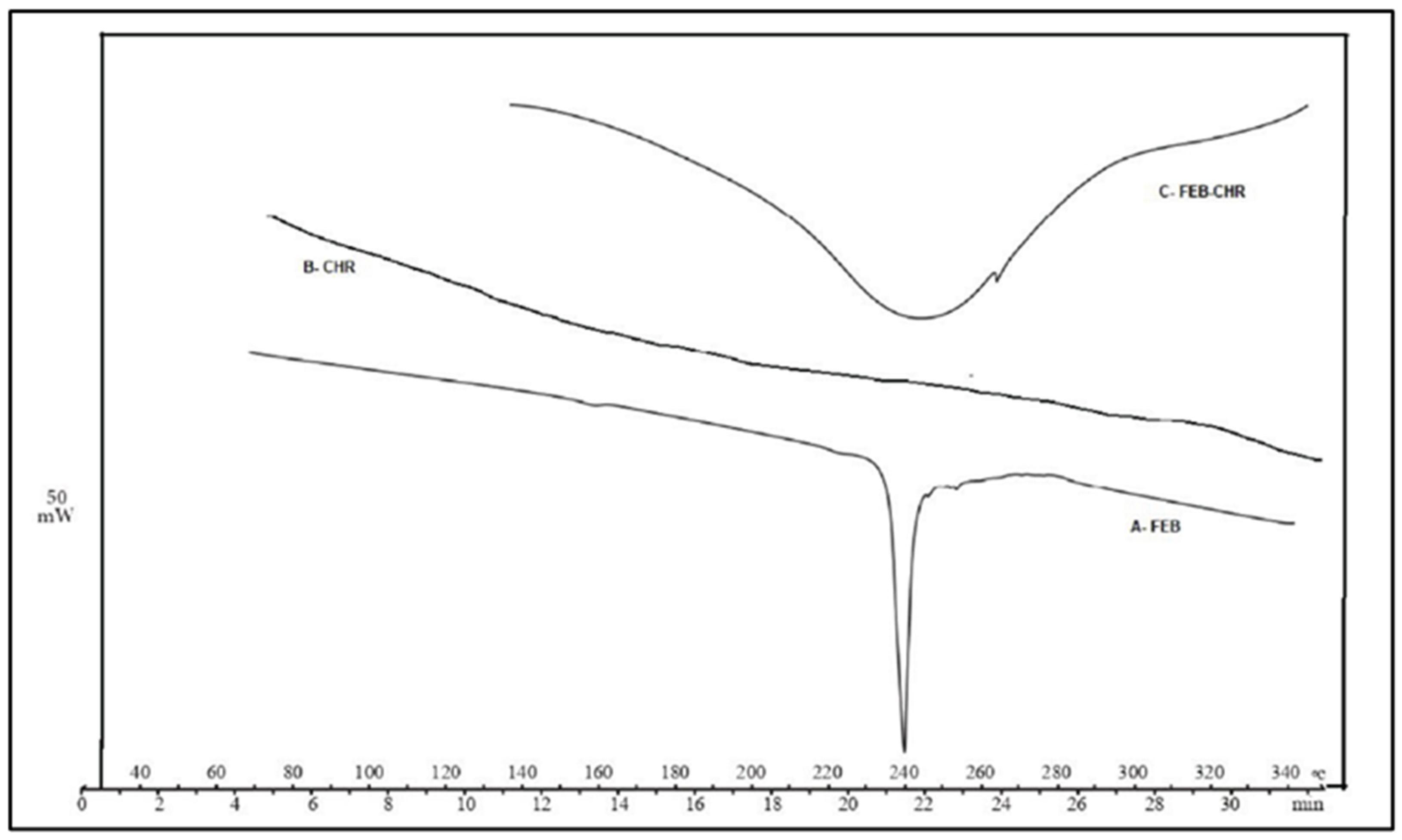
2.2. Evaluation of the FEB-CHR Mixture
2.3. Flowability and Compressibility
2.4. Evaluation of the Tablets
2.5. Dissolution and Drug Release
2.6. In Vivo Pharmacokinetic Study
3. Discussion
3.1. Pharmacokinetic Parameter Comparison
- Cmax (maximum concentration):
- 2.
- Tmax (time to reach Cmax):
- 3.
- AUC (area under the curve):
3.2. Consistency and Correlations between In Vitro and In Vivo Outcomes
4. Materials and Methods
4.1. Materials
4.2. HPLC Method of Analysis of FEB
4.3. Preparation of the Tablets
4.4. Evaluation of the FEB-CHR Mixture
4.4.1. Fourier-Transform Infrared (FTIR) Spectroscopy
4.4.2. Differential Scanning Calorimetry (DSC)
4.4.3. Particle Size Measurement
4.4.4. Adsorption Density
4.5. Evaluation of Compressibility of the Granules
4.6. Evaluation of the Prepared Tablets
4.7. In Vivo Study and Measurement of Bioavailability
4.7.1. LC-MS/MS Method of Analysis of FEB in Rats’ Plasma
4.7.2. Model Tablet Preparation
4.7.3. Measurement of Pharmacokinetic Parameters
4.7.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gerriets, V.; Patel, P.; Jialal, I. Febuxostat; StatPearls: St. Petersburg, FL, USA, 2024. [Google Scholar]
- Liu, Y.; Liang, Y.; Yuhong, J.; Xin, P.; Han, J.L.; Zhu, R.; Zhang, M.; Chen, W.; Ma, Y.; Du, Y.; et al. Advances in Nanotechnology for Enhancing the Solubility and Bioavailability of Poorly Soluble Drugs. Drug Des. Dev. Ther. 2024, 18, 1469. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 848043. [Google Scholar] [CrossRef]
- Reinders, M.K.; Jansen, T.L.T.A. Management of Hyperuricemia in Gout: Focus on Febuxostat. Clin. Interv. Aging 2010, 5, 7. [Google Scholar] [CrossRef]
- Patel, A.M.; Patel, S.R. Enhancing Solubility of Polymer-Loaded Febuxostat through Ultrasound-Assisted Microfluidic Antisolvent Nanoprecipitation: Optimization Using Box-Behnken Design. Chem. Eng. Process.—Process Intensif. 2024, 201, 109802. [Google Scholar] [CrossRef]
- Miriyala, N.; Ouyang, D.; Perrie, Y.; Lowry, D.; Kirby, D.J. Activated Carbon as a Carrier for Amorphous Drug Delivery: Effect of Drug Characteristics and Carrier Wettability. Eur. J. Pharm. Biopharm. 2017, 115, 197–205. [Google Scholar] [CrossRef]
- Nazarkina, Z.K.; Savostyanova, T.A.; Chelobanov, B.P.; Romanova, I.V.; Simonov, P.A.; Kvon, R.I.; Karpenko, A.A.; Laktionov, P.P. Activated Carbon for Drug Delivery from Composite Biomaterials: The Effect of Grinding on Sirolimus Binding and Release. Pharmaceutics 2022, 14, 1386. [Google Scholar] [CrossRef]
- Wong, C.F.; Saif, U.M.; Chow, K.L.; Wong, J.T.F.; Chen, X.W.; Liang, Y.; Cheng, Z.; Tsang, Y.F.; Wong, M.H.; Man, Y.B. Applications of Charcoal, Activated Charcoal, and Biochar in Aquaculture—A Review. Sci. Total Environ. 2024, 929, 172574. [Google Scholar] [CrossRef]
- Joshi, P.B.; Chavali, M.; Tripati, G.K.; Tondwalkar, S. Medical Applications of Activated Carbon. In Activated Carbon; The Royal Society of Chemistry: London, UK, 2023; pp. 42–54. [Google Scholar] [CrossRef]
- Hamed, R. Physiological Parameters of the Gastrointestinal Fluid Impact the Dissolution Behavior of the BCS Class IIa Drug Valsartan. Pharm. Dev. Technol. 2018, 23, 1168–1176. [Google Scholar] [CrossRef]
- Sohn, J.S.; Choi, J.S. Development and Evaluation of Febuxostat Solid Dispersion through Screening Method. Saudi Pharm. J. 2023, 31, 101724. [Google Scholar] [CrossRef]
- Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/021856s000toc.cfm (accessed on 20 August 2024).
- Febuxostat|C16H16N2O3S|CID 134018—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Febuxostat (accessed on 20 August 2024).
- Malik, N.; Dhiman, P.; Khatkar, A. In Silico Design and Synthesis of Targeted Rutin Derivatives as Xanthine Oxidase Inhibitors. BMC Chem. 2019, 13, 71. [Google Scholar] [CrossRef]
- Sánchez-Lozada, L.G.; Tapia, E.; Bautista-García, P.; Soto, V.; Ávila-Casado, C.; Vega-Campos, I.P.; Nakagawa, T.; Zhao, L.; Franco, M.; Johnson, R.J. Effects of Febuxostat on Metabolic and Renal Alterations in Rats with Fructose-Induced Metabolic Syndrome. Am. J. Physiol. Renal Physiol. 2008, 294, F710–F718. [Google Scholar] [CrossRef]
- Jairoun, A.A.; Al-Hemyari, S.S.; Shahwan, M.; Hassan, N.; Zyoud, S.H.; Jaber, A.A.S.; Al-Qirim, T. Insights into Metformin XR Pharmacotherapy Knowledge Among Community Pharmacists: A Cross-Sectional Study. Clin. Med. Insights Endocrinol. Diabetes 2023, 16, 11795514231203913. [Google Scholar] [CrossRef]
- Creteanu, A.; Lisa, G.; Vasile, C.; Popescu, M.C.; Pamfil, D.; Lungu, C.N.; Panainte, A.D.; Tantaru, G. New Hydrophilic Matrix Tablets for the Controlled Released of Chlorzoxazone. Int. J. Mol. Sci. 2024, 25, 5137. [Google Scholar] [CrossRef]
- Zalte, H.D.; Saudagar, R.B. Review on sustained release matrix tablet. Int. J. Pharm. Biol. Sci. 2013, 3, 17–29. [Google Scholar]
- Ogueri, K.S.; Shamblin, S.L. Osmotic-Controlled Release Oral Tablets: Technology and Functional Insights. Trends Biotechnol. 2022, 40, 606–619. [Google Scholar] [CrossRef]
- Kumar, V.; Somkuwar, S.; Singhai, A.K.; Kumar, V.; Somkuwar, S.; Singhai, A.K. A Recent Update on Gastro Retentive Drug Delivery Systems. GSC Biol. Pharm. Sci. 2024, 27, 125–144. [Google Scholar] [CrossRef]
- Vrettos, N.N.; Roberts, C.J.; Zhu, Z. Gastroretentive Technologies in Tandem with Controlled-Release Strategies: A Potent Answer to Oral Drug Bioavailability and Patient Compliance Implications. Pharmaceutics 2021, 13, 1591. [Google Scholar] [CrossRef]
- Tayyab, M.; Haseeb, M.T.; Alsahli, T.G.; Khaliq, N.U.; Hussain, M.A.; Khan, R.; Nawaz, A.; Iqbal, A.; Alanazi, A.S.; Bukhari, S.N.A. Fabrication and Optimization of Febuxostat-Loaded Chitosan Nanocarriers for Better Pharmacokinetics Profile. Int. J. Biol. Macromol. 2024, 257, 128448. [Google Scholar] [CrossRef]
- Ahuja, B.K.; Jena, S.K.; Paidi, S.K.; Bagri, S.; Suresh, S. Formulation, Optimization and in Vitro-in Vivo Evaluation of Febuxostat Nanosuspension. Int. J. Pharm. 2015, 478, 540–552. [Google Scholar] [CrossRef]
- Rathi, S.; Vaghela, S.; Shah, R.; Shah, S. Formulation and Evaluation of Gastroretentive Floating Tablets of Febuxostat. Res. J. Pharm. Technol. 2021, 14, 5359–5365. [Google Scholar] [CrossRef]
- Sohn, J.S.; Choi, J.S. Febuxostat Solubilization and Stabilization Approach Using Solid Dispersion Method: Synergistic Effect of Dicalcium Phosphate Dehydrate and Chitosan. Int. J. Biol. Macromol. 2023, 253, 127266. [Google Scholar] [CrossRef]
- Pan, Y.; Sahoo, N.G.; Li, L. The Application of Graphene Oxide in Drug Delivery. Expert Opin. Drug Deliv. 2012, 9, 1365–1376. [Google Scholar] [CrossRef]
- Wheless, J.W.; Phelps, S.J. A Clinician’s Guide to Oral Extended-Release Drug Delivery Systems in Epilepsy. J. Pediatr. Pharmacol. Ther. JPPT 2018, 23, 277. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, H.; Kim, T.E.; Lee, S.H.; Chee, D.H.; Cho, J.Y.; Yu, K.S.; Jang, I.J. Comparative Steady-State Pharmacokinetic Study of an Extended-Release Formulation of Itopride and Its Immediate-Release Reference Formulation in Healthy Volunteers. Drug Des. Dev. Ther. 2014, 8, 123. [Google Scholar] [CrossRef]
- Boaretto, E.; Rebollo, N.R.; Cohen-Ofri, I.; Popovitz-Biro, R.; Bar-Yosef, O.; Meignen, L.; Goldberg, P.; Weiner, S. Structural Characterization of Charcoal Exposed to High and Low Ph: Implications for 14C Sample Preparation and Charcoal Preservation. Radiocarbon 2008, 50, 289–307. [Google Scholar] [CrossRef]
- Nitayaphat, W.; Jiratumnukul, N.; Charuchinda, S.; Kittinaovarat, S. Mechanical Properties of Chitosan/Bamboo Charcoal Composite Films Made with Normal and Surface Oxidized Charcoal. Carbohydr. Polym. 2009, 78, 444–448. [Google Scholar] [CrossRef]
- Febuxostat: Uses, Interactions, Mechanism of Action|DrugBank Online. Available online: https://go.drugbank.com/drugs/DB04854 (accessed on 20 August 2024).
- Majeed, B.J.M.; Saadallah, M.A.; Al-Ani, I.H.; El-Tanani, M.K.; Al Azzam, K.M.; Abdallah, H.H.; Al-Hajji, F. Evaluation of Solubility, Dissolution Rate, and Oral Bioavailability of β-Cyclodextrin and Hydroxypropyl β-Cyclodextrin as Inclusion Complexes of the Tyrosine Kinase Inhibitor, Alectinib. Pharmaceuticals 2024, 17, 737. [Google Scholar] [CrossRef]
- Drug Adsorption—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/drug-adsorption (accessed on 14 September 2024).
- Coelho, J.F.; Ferreira, P.C.; Alves, P.; Cordeiro, R.; Fonseca, A.C.; Góis, J.R.; Gil, M.H. Drug Delivery Systems: Advanced Technologies Potentially Applicable in Personalized Treatments. EPMA J. 2010, 1, 164. [Google Scholar] [CrossRef]
- Adepu, S.; Ramakrishna, S. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef]
- Herdiana, Y.; Wathoni, N.; Shamsuddin, S.; Muchtaridi, M. Drug Release Study of the Chitosan-Based Nanoparticles. Heliyon 2022, 8, e08674. [Google Scholar] [CrossRef]
- Kranz, H.; Yilmaz, E.; Brazeau, G.A.; Bodmeier, R. In Vitro and in Vivo Drug Release from a Novel in Situ Forming Drug Delivery System. Pharm. Res. 2008, 25, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Samala, A.; Pawar, S.; Manala, S.; Chada, S.; Nageshwar, M. Development and Validation of RP-HPLC Method for Estimation of Risperidone in Tablet Dosage Form. Res. J. Pharm. Technol. 2013, 6, 659–661. [Google Scholar]
- ANALYTICAL PROCEDURE DEVELOPMENT Q14. Available online: https://database.ich.org/sites/default/files/ICH_Q14_Document_Step2_Guideline_2022_0324.pdf (accessed on 20 August 2024).
- Mestre, A.S.; Pires, J.; Nogueira, J.M.F.; Carvalho, A.P. Activated Carbons for the Adsorption of Ibuprofen. Carbon 2007, 45, 1979–1988. [Google Scholar] [CrossRef]
- Chang, E.E.; Wan, J.C.; Kim, H.; Liang, C.H.; Dai, Y.D.; Chiang, P.C. Adsorption of Selected Pharmaceutical Compounds onto Activated Carbon in Dilute Aqueous Solutions Exemplified by Acetaminophen, Diclofenac, and Sulfamethoxazole. Sci. World J. 2015, 2015, 186501. [Google Scholar] [CrossRef]
- Yanan, C.; Srour, Z.; Ali, J.; Guo, S.; Taamalli, S.; Fèvre-Nollet, V.; da Boit Martinello, K.; Georgin, J.; Franco, D.S.P.; Silva, L.F.O.; et al. Adsorption of Paracetamol and Ketoprofenon Activated Charcoal Prepared from the Residue of the Fruit of Butiacapitate: Experiments and Theoretical Interpretations. Chem. Eng. J. 2023, 454, 139943. [Google Scholar] [CrossRef]
- Bacher, C.; Olsen, P.M.; Bertelsen, P.; Sonnergaard, J.M. Compressibility and Compactibility of Granules Produced by Wet and Dry Granulation. Int. J. Pharm. 2008, 358, 69–74. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Code | Ratio of FEB:CHR | The Angle of Repose (θ) | Carr’s Index | Hausner Ratio | Flowability Description |
|---|---|---|---|---|---|
| F1 | 1:2 | 36 ± 1.0 | 18.2 ± 1.0 | 1.18 ± 0.05 | fair |
| F2 | 1:1.75 | 35 ± 1.5 | 17.0 ± 0.5 | 1.15 ± 0.03 | good |
| F3 | 1:1.5 | 34 ± 2.0 | 13.0 ± 0.5 | 1.13 ± 0.02 | good |
| F4 | 1:1.25 | 33 ± 1.0 | 13.0 ±0.9 | 1.12 ± 0.05 | good |
| F5 | 1:1 | 33 ± 0.75 | 12.8 ± 1.0 | 1.11 ± 0.04 | good |
| F6 | 1:0.75 | 32 ± 1.0 | 12.5 ±1.0 | 1.11 ± 0.03 | good |
| F7 | 1:0.5 | 31 ± 0.6 | 11.5 ±1.0 | 1.11 ± 0.09 | good |
| F8 | 1:0.25 | 31 ± 0.5 | 11.2 ± 0.6 | 1.10 ± 0.05 | good |
| Formula Code | Ratio of FEB:CHR | Weight Uniformity (mg ± SD) N = 20 | Hardness (Kg/cm2) N = 3 | Friability (%) | Disintegration Time (min) N = 6 |
|---|---|---|---|---|---|
| F1 | 1:2 | 397 ± 2 | 3.5 ± 1.2 | 1.3 | 2.5 ± 0.5 |
| F2 | 1:1.75 | 398 ± 1.5 | 3.6 ± 1.3 | 1.2 | 2.6 ± 0.6 |
| F3 | 1:1.5 | 401 ± 2 | 3.9 ± 0.95 | 1.25 | 3.0 ± 1.0 |
| F4 | 1:1.25 | 400 ± 2.3 | 4.0 ± 1.5 | 1.0 | 3.2 ± 1.0 |
| F5 | 1:1 | 398 ± 2.1 | 5.2 ± 1.8 | 1.0 | 4.1 ± 1.2 |
| F6 | 1:0.75 | 402 ±1.6 | 6.1 ± 1.9 | 0.78 | 4.0 ± 0.8 |
| F7 | 1:0.5 | 403 ± 2.3 | 7.0 ± 1.0 | 0.65 | 4.0 ± 0.9 |
| F8 | 1:0.25 | 401 ± 1.9 | 7.5 ± 1.2 | 0.65 | 4.5 ± 0.75 |
| Mode/Formula Code | Zero-Order (Cum % Drug Release vs. Time) | First-Order (Log (Cum % Drug Release vs. Time) | Peppas Model (Log (Cum % Drug Release vs. Time) | Higuchi Model (Cum % Drug Release vs. SQRT of Time) | Hixson–Crowell Model W0 (1/3)–Wt (1/3) vs. Time |
|---|---|---|---|---|---|
| F1 | 0.835 | 0.841 | 0.972 | 0.941 | 0.835 |
| F2 | 0.794 | 0.797 | 0.962 | 0.914 | 0.793 |
| F3 | 0.746 | 0.749 | 0.935 | 0.873 | 0.746 |
| F4 | 0.969 | 0.975 | 0.990 | 0.983 | 0.969 |
| F5 | 0.972 | 0.980 | 0.996 | 0.989 | 1.00 |
| F6 | 0.972 | 0.990 | 0.979 | 0.989 | 0.972 |
| F7 | 0.994 | 0.995 | 0.987 | 0.970 | 0.994 |
| F8 | 0.986 | 0.977 | 0.986 | 0.968 | 0.999 |
| The Formula | Cmax (ng/mL) | AUC 0–12 (ng·mL/h) | AUC Total (ng·mL/h) | Tmax (h) |
|---|---|---|---|---|
| Reference Formula | 150 ± 25 | 665.5 ± 120 | 673.04 ± 180 | 2.00 ± 1.0 |
| F8 | 155 ± 38 | * 1134.5 ± 200 | * 1294.24 ± 320 | 3.00 ± 0.5 |
| Formula Code | Ratio of FEB:CHR | FEB (mg) | CHR (mg) | Avecil 102 (mg) | Sodium Starch Glycolate (mg) | Magnesium Stearate (mg) | Total Weight of the Tablet |
|---|---|---|---|---|---|---|---|
| F1 | 1:2 | 80 | 160 | 146 | 10 | 4 | 400 |
| F2 | 1:1.75 | 80 | 140 | 166 | 10 | 4 | 400 |
| F3 | 1:1.5 | 80 | 120 | 186 | 10 | 4 | 400 |
| F4 | 1:1.25 | 80 | 100 | 206 | 10 | 4 | 400 |
| F5 | 1:1 | 80 | 80 | 174 | 10 | 4 | 400 |
| F6 | 1:0.75 | 80 | 60 | 246 | 10 | 4 | 400 |
| F7 | 1:0.5 | 80 | 40 | 266 | 10 | 4 | 400 |
| F8 | 1:0.25 | 80 | 20 | 286 | 10 | 4 | 400 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Ani, I.H.; Hailat, M.; Mohammed, D.J.; Matalqah, S.M.; Abu Dayah, A.A.; Majeed, B.J.M.; Awad, R.; Filip, L.; Abu Dayyih, W. Development and Evaluation of a Cost-Effective, Carbon-Based, Extended-Release Febuxostat Tablet. Molecules 2024, 29, 4629. https://doi.org/10.3390/molecules29194629
Al-Ani IH, Hailat M, Mohammed DJ, Matalqah SM, Abu Dayah AA, Majeed BJM, Awad R, Filip L, Abu Dayyih W. Development and Evaluation of a Cost-Effective, Carbon-Based, Extended-Release Febuxostat Tablet. Molecules. 2024; 29(19):4629. https://doi.org/10.3390/molecules29194629
Chicago/Turabian StyleAl-Ani, Israa Hamid, Mohammad Hailat, Dina J. Mohammed, Sina Mahmoud Matalqah, Alaa Azeez Abu Dayah, Bashar J. M. Majeed, Riad Awad, Lorena Filip, and Wael Abu Dayyih. 2024. "Development and Evaluation of a Cost-Effective, Carbon-Based, Extended-Release Febuxostat Tablet" Molecules 29, no. 19: 4629. https://doi.org/10.3390/molecules29194629