1. Introduction
Primary osteosarcoma is the most common bone tumor, and occurs predominantly in adolescents and young adults [
1]. Even after the introduction of aggressive chemotherapy and wide excision of tumors, 30–50% of patients with initially localized disease subsequently develop recurrence, with subsequently poor clinical outcomes. Moreover, 20–30% of newly diagnosed cases present with metastatic disease [
2,
3]. The identification of the effector molecules and/or signal transduction pathways responsible for regulating carcinogenesis and malignant development is therefore crucial for understanding and isolating potential molecular targets that could be used to disrupt the tumor machinery, whilst protecting the integrity and function of normal tissue.
Akt is a serine/threonine kinase that plays a central role in tumorigenesis. Among the members of Akt family, Akt2 is associated with the development of human cancers. Recent reports have demonstrated that the PI3K/Akt pathway is a potent survival signal that may mediate resistance to the apoptotic effects of chemotherapy and radiation therapy in a variety of cancer types [
4–
6].
p53 up-regulated modulator of apoptosis (PUMA) is an essential mediator of cell death and plays a key functional role in the process of p53-mediated apoptosis [
7,
8]. PUMA activation was recently reported to be a mechanism for the augmentation of cisplatin cytotoxicity in QBC939 cells [
9]. Alison
et al. [
10] recently found that Akt2 inhibition enhanced the inhibitory effects of PUMA on melanoma cell survival
in vitro and on melanoma tumor growth
in vivo.
In this study, we investigated the hypothesis that knockdown of Akt2 plays an important role in osteosarcoma cell chemosensitivity to cisplatin-induced apoptosis through upregulation of PUMA.
2. Materials and Methods
2.1. Cell Culture
The human osteosarcoma cell line SAOS-2 was obtained from the American Type Culture Collection (Manassas, VA, USA) and was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U/mL) and streptomycin (100 U/mL) at 37 °C in an atmosphere of 5% CO2.
2.2. Immunohistochemistry
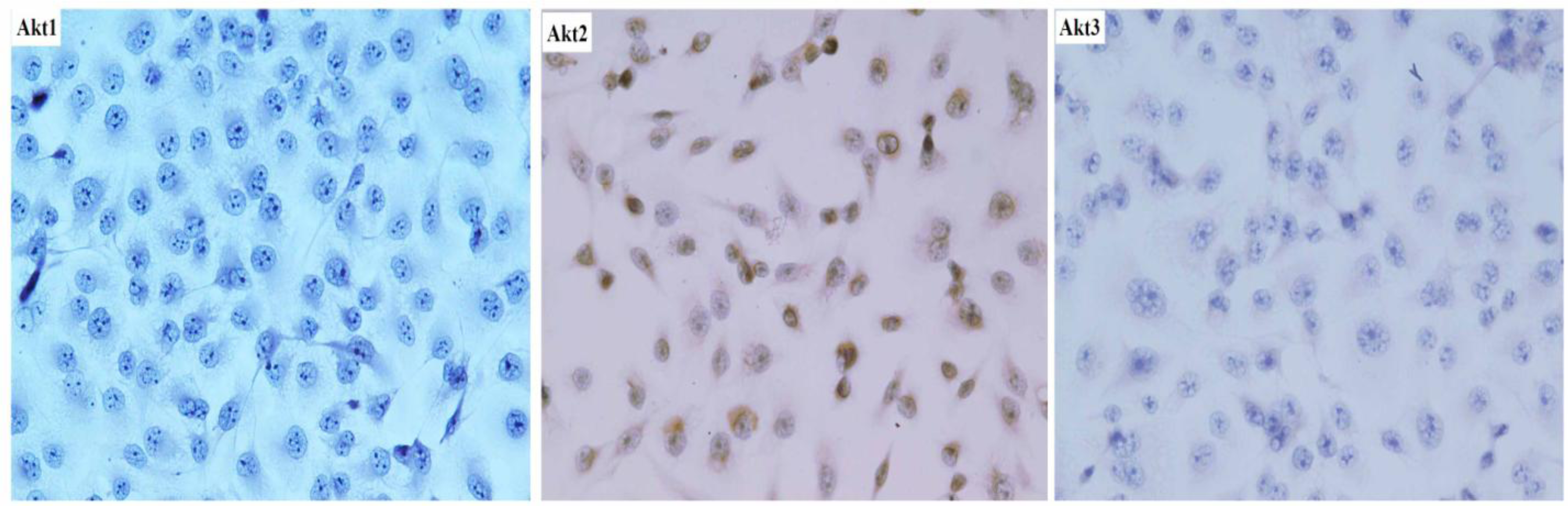
SAOS-2 cells were plated at a density of 5 × 104/mm2 in culture dishes for 24 h. After incubation, dishes were washed twice with cold saline. Subsequently, the cells in the dishes were fixed in 0.3 mol/L NaCl in 70% ethanol for 30 min. The fixed cells were lysed in 0.12 N HCl in 70% ethanol for 10 min and washed with PBS for 5 min three times. The cells were incubated with 1% normal horse serum in PBS for 30 min at room temperature. After washes with PBS for 5 min, Anti-Akt1 (B-1)(1:100, Santa Cruz), Anti-Akt2 (1:100, Cell Signaling Technology, Inc., Beverly, MA, USA) and Anti-Akt3 (M-14) (1:100, Cell Signaling Technology) was used as the primary antibody, followed by the rabbit ABC staining system. Protein-positive cells were counted by light microscopy at ×200 magnification and expressed as the number of positive cells per 1000 cancer cells.
2.3. Small Interference (siRNA) Transfection
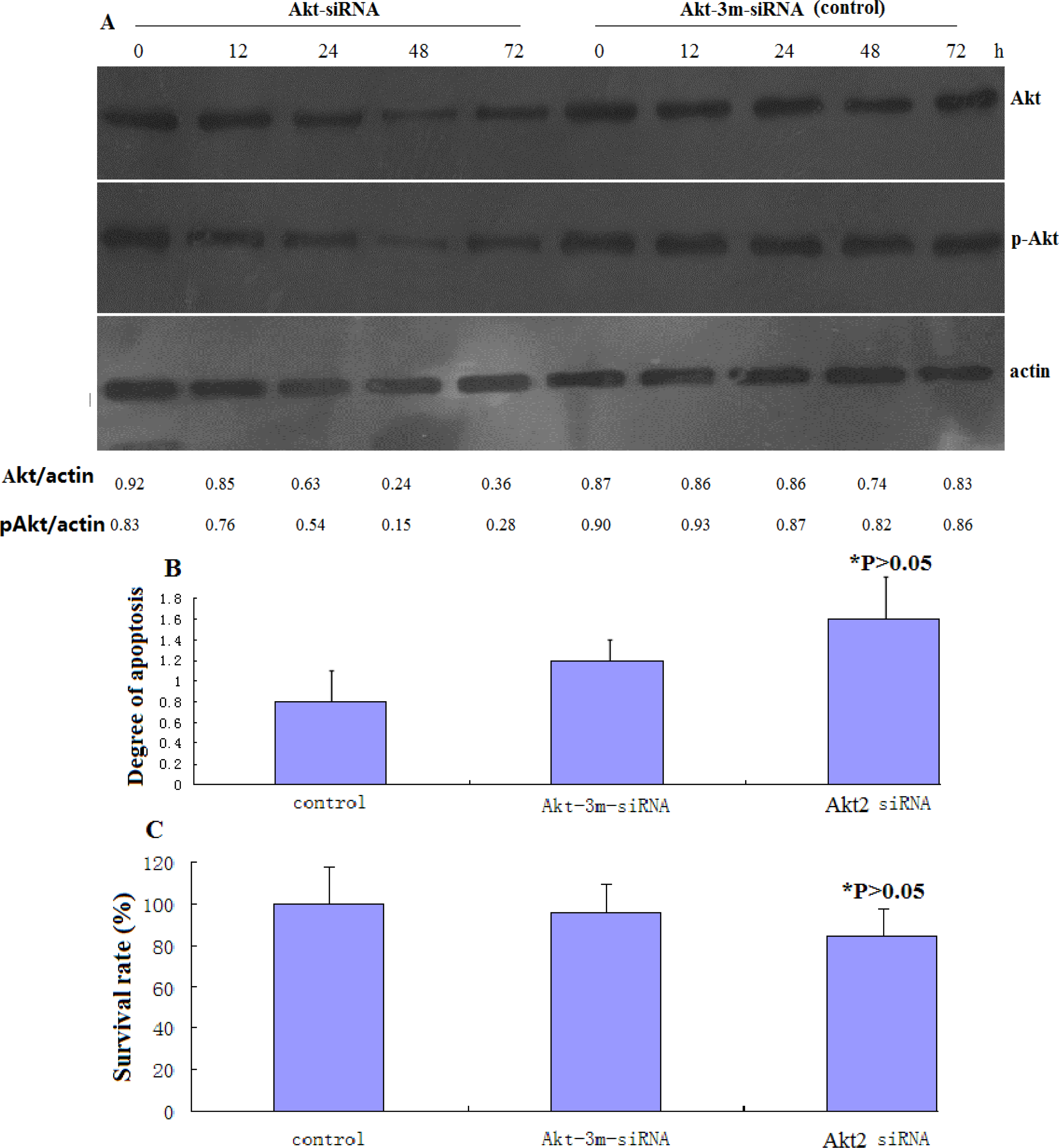
The following siRNA sequences were utilized in this study: Akt2-siRNA (sense 5′-UGCCCUUCUACAACCAGGAdTdT-3′), Akt-3m-siRNA (control siRNA for transfection) (sense 5′-UGCCGUUCUUCAACGAGGAdTdT-3′) [
11]. The siRNA oligonucleotides, together with the corresponding antisense oligonucleotides, were synthesized by Dharmacon (Lafayette, CO, USA). The control siRNA was from Ambion (Austin, TX, USA). siRNA transfection was performed using Lipofectamine 2000, according to the manufacturer's instructions, and following procedures described previously [
12]. Pilot experiments were performed to optimize the amount and time of maximal protein knockdown. Cells were treated with cisplatin following siRNA transfection, during the period of maximal protein knockdown.
2.4. Cell Proliferation and Cytotoxicity Assay
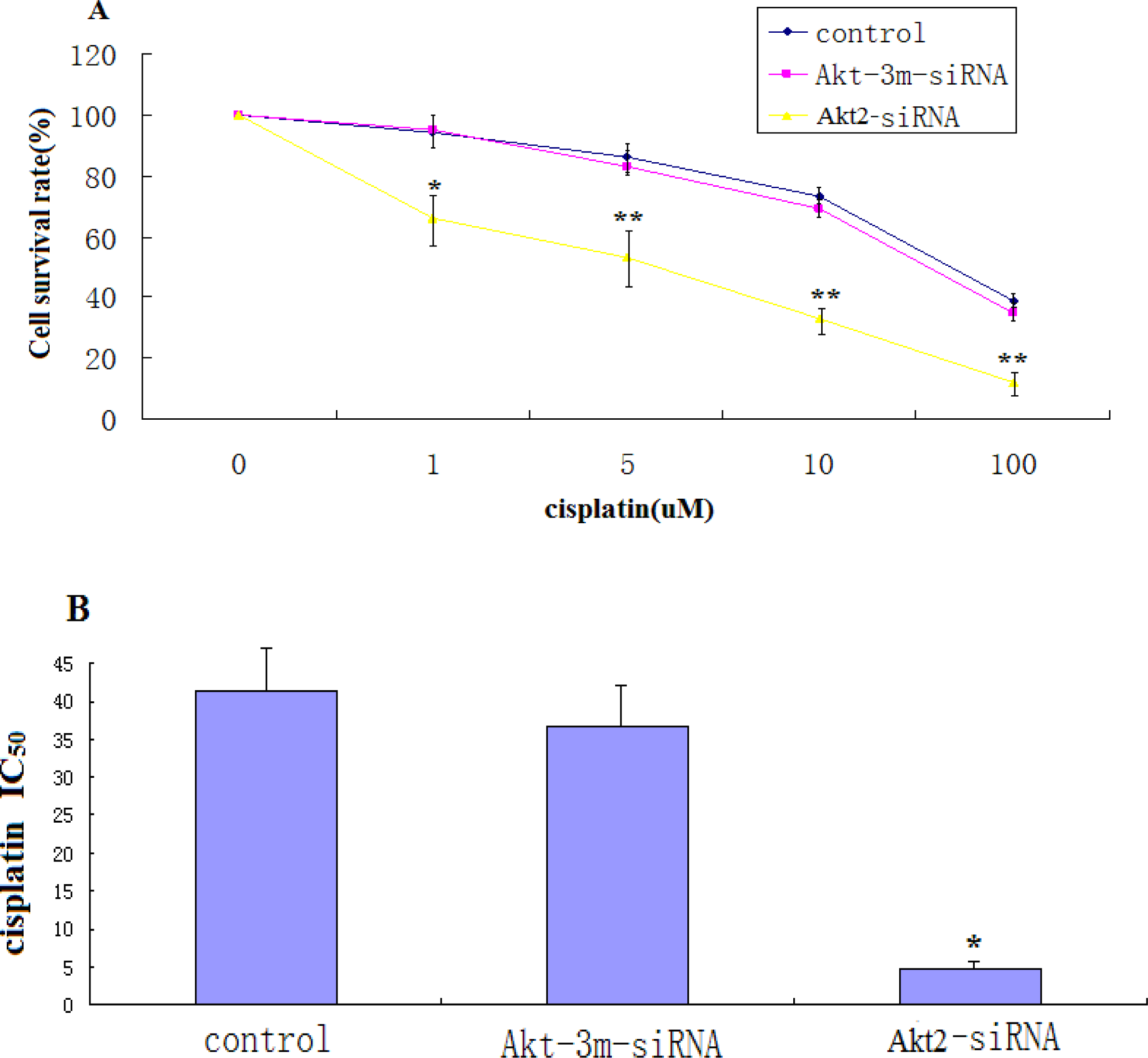
The effects of Akt2 silencing on growth of SAOS-2 cells were assessed by culturing control cells and cells transfected with Akt2-siRNA in the presence of 1 μg/mL cisplatin for 48 h. Cells from these precultures were seeded at 3 × 103 cells/well into 96-well plates and allowed to attach for 12 h. Growth was then measured by determining the numbers of living cells using the MTT method. The results are expressed as percentage of viable cells at the start of the experiment.
For cytotoxicity assays, 5 × 103 cells/well were plated in 96-well plates and incubated overnight. The medium was then replaced with fresh medium containing different concentrations of cisplatin (Eli Lilly, Geneva, Switzerland) and incubation was continued for 48 h. Cisplatin (1 μg/mL) was present throughout the experiment. Cell viability was determined using the MTT method. Percent survival was defined as 100 × (T − T0)/(C − T0) when (T − T0) = 0. When the T value was less than T0, cell killing had occurred and the cytotoxic activity was determined as (T − T0)/T0 expressed as a percentage. T is the optical density (OD)540 value at the time-point in question, and T0 is the OD540 value at the moment of drug addition. C indicates the OD540 value of the untreated control group at the time-point in question. IC50 values were calculated from three independent experiments.
2.5. Apoptosis Detection by Enzyme-Linked Immunosorbent Assay (ELISA)
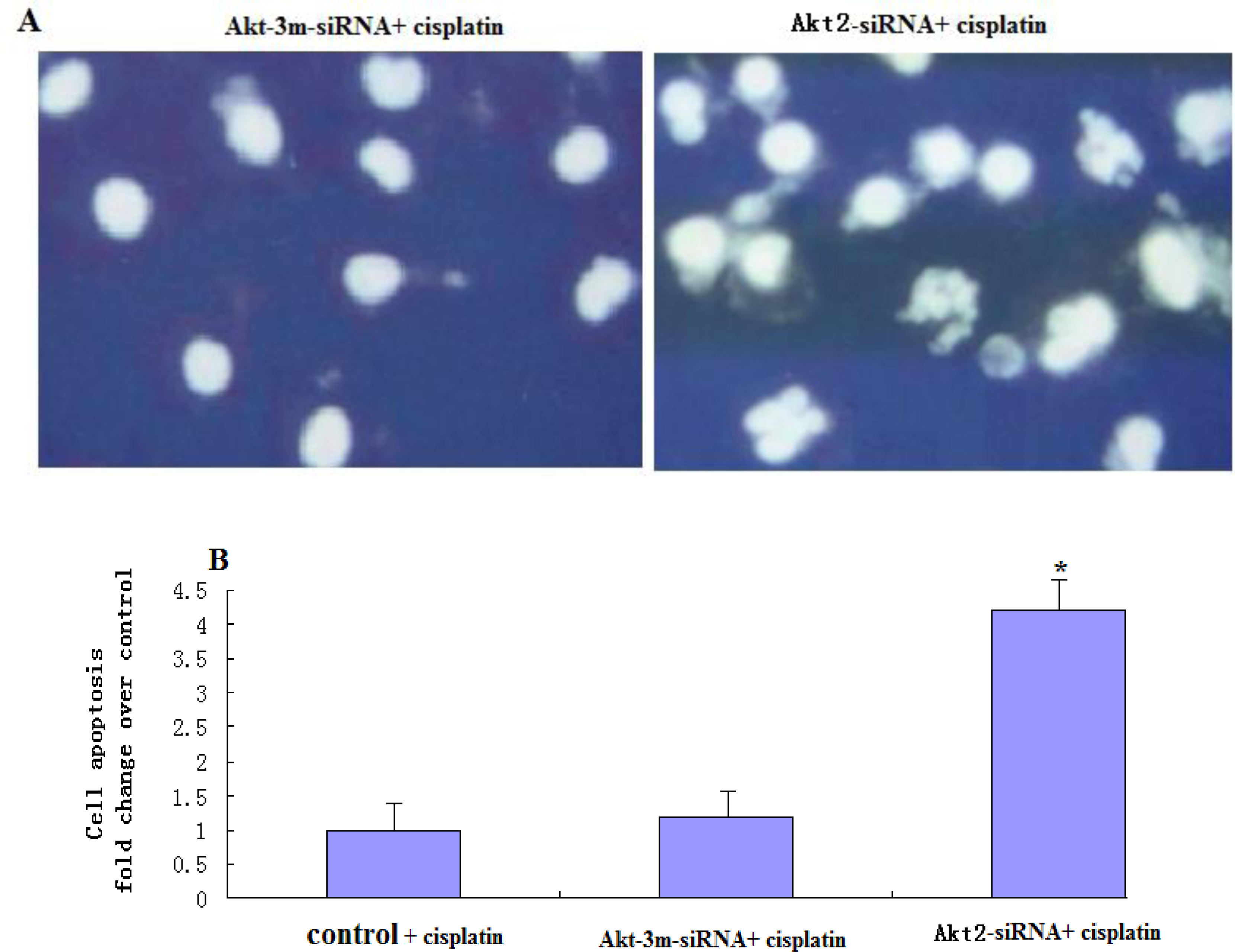
This assay was performed using the cell death detection ELISAPLUS kit (Roche Applied Sciences, Indianapolis, IN, USA), according to the manufacturer’s instructions. Cells of both transfected and untransfected cells were plated on 6-well plates and treated with different indicated concentrations of cisplatin, after which both adherent and floating (apoptotic) populations were harvested. Cells were lysed in NP-40 lysis buffer and nucleosomes in the supernatant were detected photometrically using an ELISA plate Reader (SpectraMax 190, Molecular Devices Ca). The readings were expressed as degree of apoptosis relative to the untreated control, which was scored as 1.
2.6. Analysis of Apoptosis by DAPI Staining
For apoptosis analysis, both adherent and non-adherent cells were harvested after cisplatin treatment and their cell morphologies were examined by microscopy. DNA damage characteristic of apoptosis was identified by staining with 4′,6-diamidino-2-phenylindole (DAPI). Briefly, cells were harvested, washed, and fixed in 3.7% formaldehyde at room temperature for 15 min. After treatment with RNase A, samples were stained with 1 μg/mL of DAPI (Sigma USA) in phosphate-buffered saline (PBS) for 15 min at room temperature, rinsed in PBS, and analyzed using a Leica DM RXE fluorescence microscope (Leica, Wetzlar, Germany). Apoptotic cells were defined by the condensation of nuclear chromatin, fragmentation, or margination to the nuclear membrane.
2.7. Western Blotting
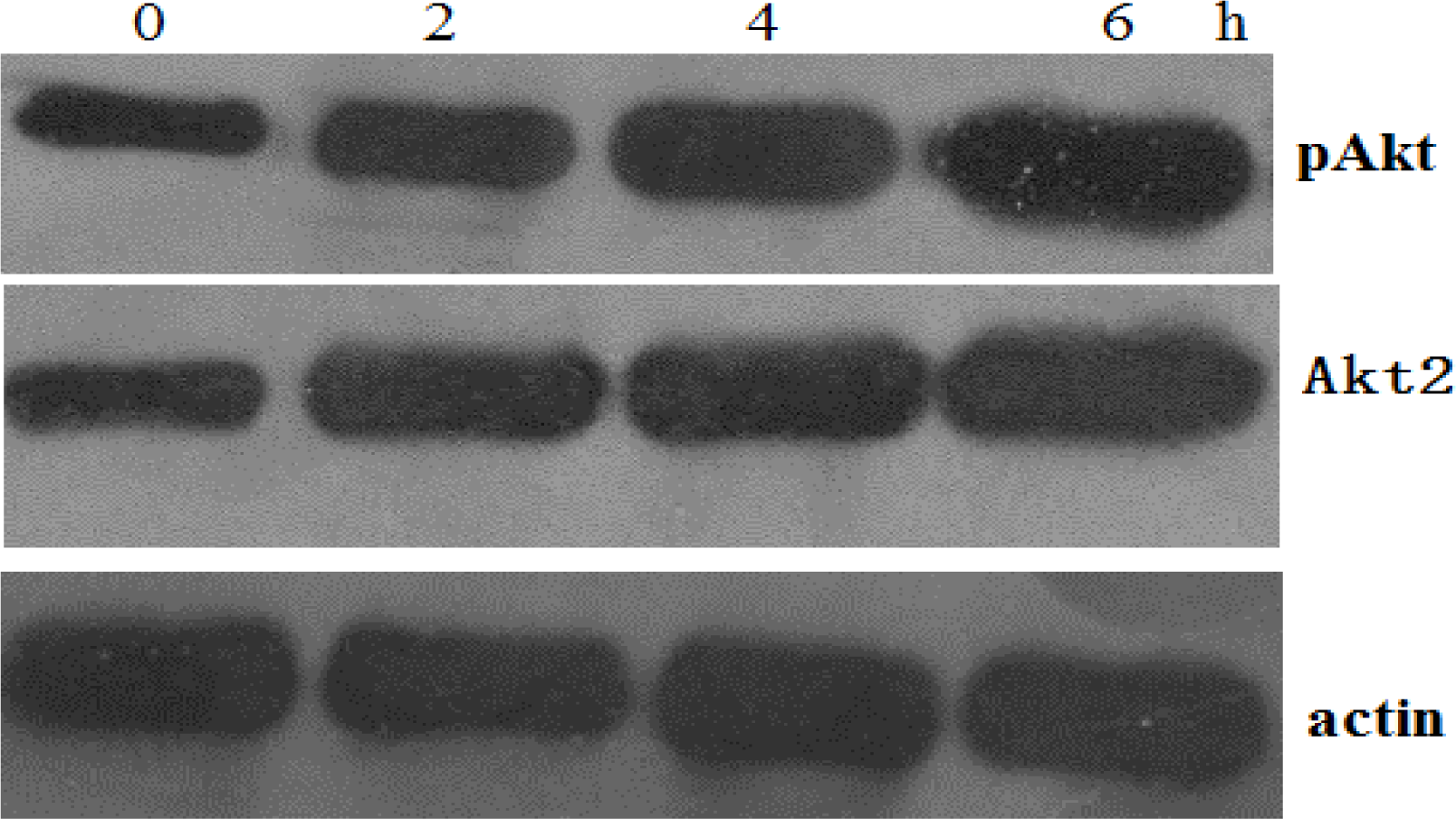
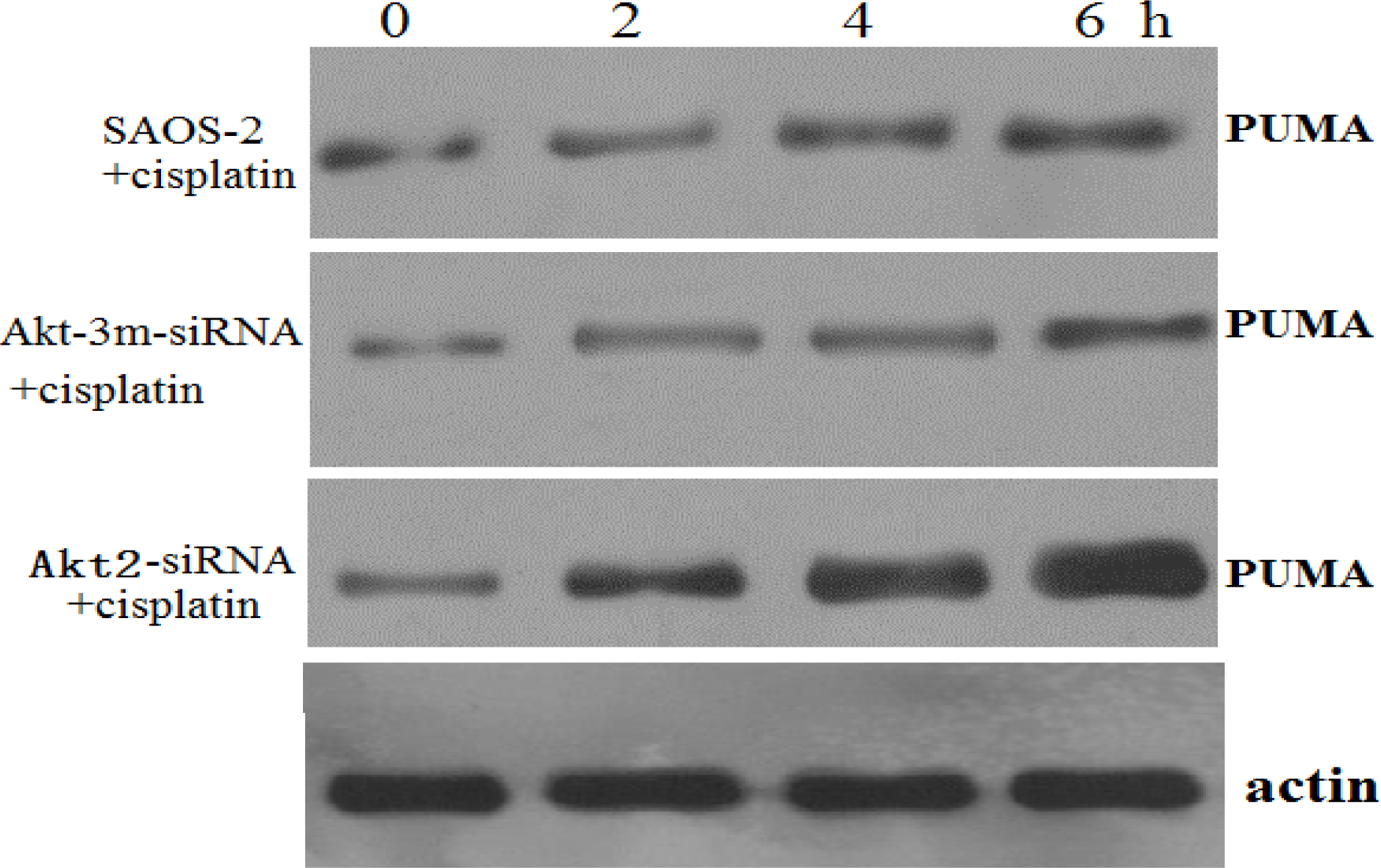
Cells were lysed in 250 mM NaCl, 50 mM Hepes (pH 7), 0.1% Nonidet P-40, and 1 μM proteinase inhibitor cocktail (Roche, Basel, Switzerland). Protein concentration was determined using the BCA protein assay (Pierce Chemical Company, Rockford, IL, USA). Equal amounts of cellular protein (20 μg/sample) were electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Zurich, Switzerland). Depending on the experiment, the membranes were first incubated with polyclonal rabbit antibody to phospho-AKT (serine-473) (Biosource International Inc., Camarillo, CA, USA), polyclonal antibody to AKT2 (Cell Signaling Technology, Inc., Beverly, MA, USA), polyclonal antibody to PUMA (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or polyclonal antibody to actin (Santa Cruz Biotechnology, Inc.). For signal detection, the blots were then incubated with peroxidase-coupled goat anti-rabbit immunoglobulin (Amersham Pharmacia Biotech). Enhanced chemiluminescence (ECL, Amersham Pharmacia Biotech) reagents were used to detect the signals, according to the manufacturer’s instructions.
2.8. Wt-P53 Assay
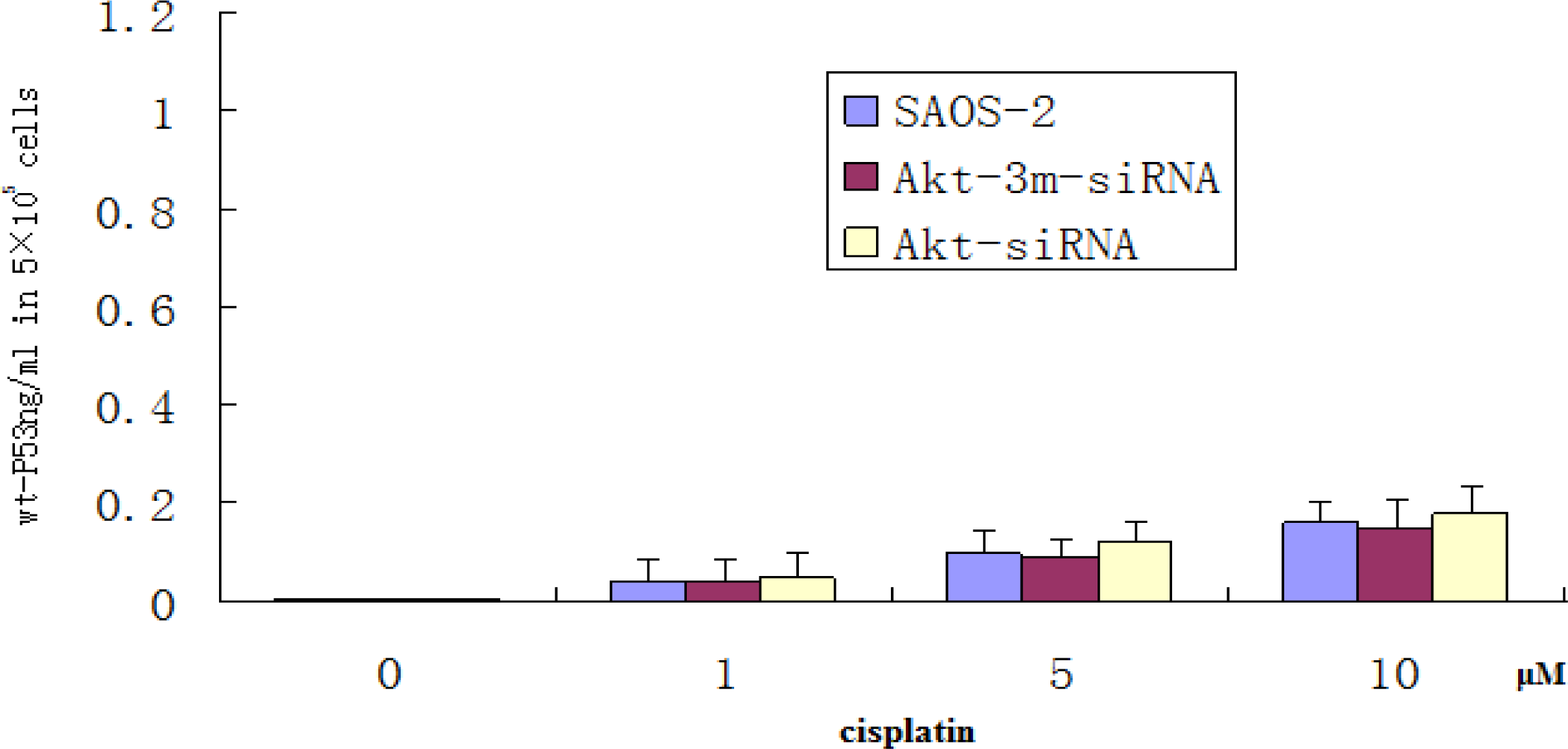
A commercial ELISA kit (Calbiochem) was used to analyze the cellular level of wt-P53. Briefly, an anti-wt-P53 monoclonal antibody (Merck-Calbiochem) was precoated onto the 96-well microtiter plate. Cells (5 × 105) of transfected and untransfected treated with different indicated concentrations of Cisplatin for 48 h was then added to the well. The plate was incubated at room temperature for 2 h and the wash was repeated. A substrate solution was then added to all wells and incubated for 30 min. At this point, a stop solution was added to all wells. Color development and intensity of the color were measured using an ELISA plate reader according to the manufacturer’s instructions. A standard curve was prepared, plotting the absorbance versus the concentration of the wt-P53 expressed as picogram per milliliter in the original samples.
2.9. Statistical Analysis
Data are presented as mean ±SD. P values were calculated using Student’s t tests or two-way analysis of variance (ANOVA) using SPSS10.0 software. P < 0.05 was considered to be statistically significant.
4. Discussion
The therapeutic advantages of cisplatin, such as high efficiency, mild side effects and easy administration, mean that it is still one of the most commonly used chemotherapeutic agents. However, resistance to cisplatin often occurs, and methods of enhancing the sensitivity of cancer cells to cisplatin-induced apoptosis have therefore become an important chemotherapeutic strategy.
The PI3K/Akt survival pathway is activated by survival signals such as growth factors, cytokines, hormones and oncogenic Ras [
14]. Activation of Akt favors survival via the direct regulation of apoptotic proteins, including the Bcl-2 members Bad and Bcl-xl or caspase 9 [
15–
18]. Akt signaling has recently been shown to mediate therapeutic resistance. The PI3K/Akt pathway is frequently overexpressed/activated in cancers, and Akt activation promotes a chemoresistant phenotype, whereas Akt inhibition sensitizes chemoresistant cells to cisplatin-induced apoptosis.
In the present study, inhibition of Akt2 had minimal effect on the basal level of apoptosis, though it reduced the threshold for the induction of apoptosis in response to chemotherapy in SAOS-2 cells. These data suggest that targeted inhibition of the Akt2 pathway may not be an adequate therapy when administered alone. However, we also examined the ability of Akt2 inhibition to sensitize SAOS-2 cells, and demonstrated increased apoptosis after combined Akt2 inhibition and cisplatin treatment, suggesting that high basal Akt2 activity is required for the efficacy of this targeted therapy. The mechanism by which Akt2 inhibition confers chemosensitivity in these cancer cells is unclear, but it appears to involve the regulation of transcription factors and proapoptotic proteins, such as PUMA. Recent evidence suggests that upregulation of PUMA is an important mechanism in cisplatin-induced apoptosis [
19], and Akt contributes to chemoresistance by attenuating p53-mediated PUMA upregulation and phosphorylation of p53, which are essential but independent determinants of cisplatin sensitivity [
20]. The results of this study showed that SAOS-2 cells responded to chemotherapy exposure with the induction of an Akt2-dependent survival pathway that may involve Akt2-mediated transcriptional inhibition of the proapoptotic PUMA gene. Akt2 inhibition enhanced the proapoptotic effect of chemotherapy via upregulating the induction of PUMA. To determine whether apoptosis induction by Akt2 knockdown and cisplatin treatment is p53 independent, ELISA method was used to detect wt-P53 level in SAOS-2 cells treated with Akt2 knockdown and/or cisplatin treatment. The results showed that although the wt-P53 level increased in a cisplatin dose dependent manner, it was not significant. It was also proven that apoptosis induction by Akt2 knockdown and cisplatin treatment is p53 independent.
Chemotherapy appears to wield a double-edged sword; although it is able to induce apoptosis in cancer cells, it is also able to stimulate the tumor cells’ native survival mechanisms. In the present study, SAOS-2 cells on the one hand responded to chemotherapy exposure by induction of an Akt2-dependent survival pathway, while on the other hand, chemotherapy exposure induced proapoptotic PUMA signaling. The benefits of combining Akt2 inhibition with standard chemotherapy thus appear to be twofold; disruption of the Akt2 survival pathway in cancer cells that harbor constitutively active Akt may induce an apoptotic response, while inhibition of Akt may abrogate the undesired survival response seen when tumor cells are exposed to chemotherapy. The chemoresistance of SAOS-2 cells may be both a manifestation of their inherent properties, but may also reflect their ability to respond to an apoptotic stimulus with the induction of a robust cell survival response that circumvents the proapoptotic effect of chemotherapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






