
Aqueous Molecular Dynamics Simulations of the M. tuberculosis Enoyl-ACP Reductase-NADH System and Its Complex with a Substrate Mimic or Diphenyl Ethers Inhibitors

 , ,
, ,
Abstract
:
1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
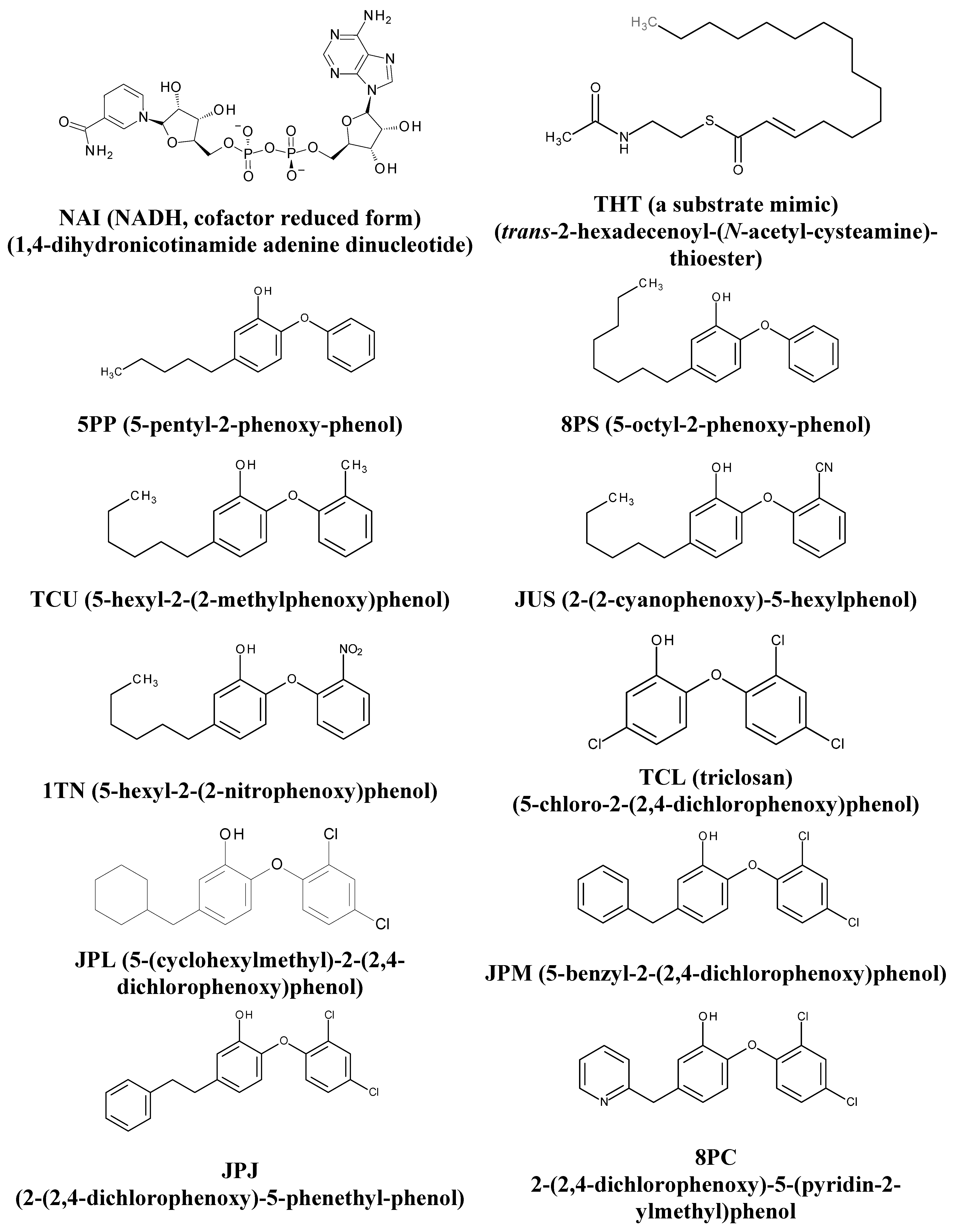
| PDB ID a | R (Å) b | Cofactor c | Ligand c | pKi (M) d | Ref. |
|---|---|---|---|---|---|
| 2AQ8 | 1.92 | NAI | n.d | n.d | [26] |
| 1BVR | 2.80 | NAD | THT | n.d | [27] |
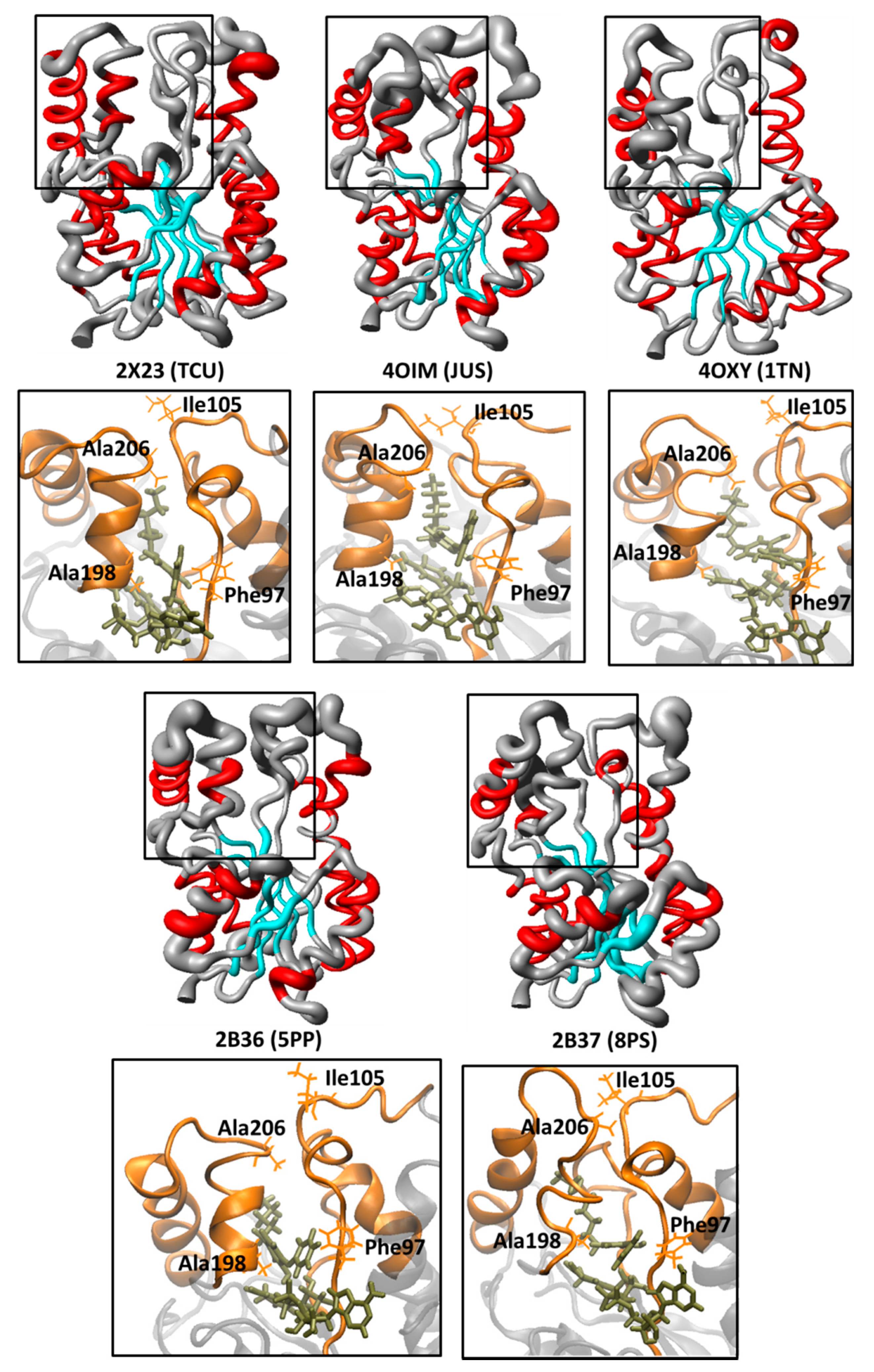
| 2X23 | 1.81 | NAD | TCU | 9.86 | [25] |
| 2B37 | 2.60 | NAD | 8PS | 8.47 | [23] |
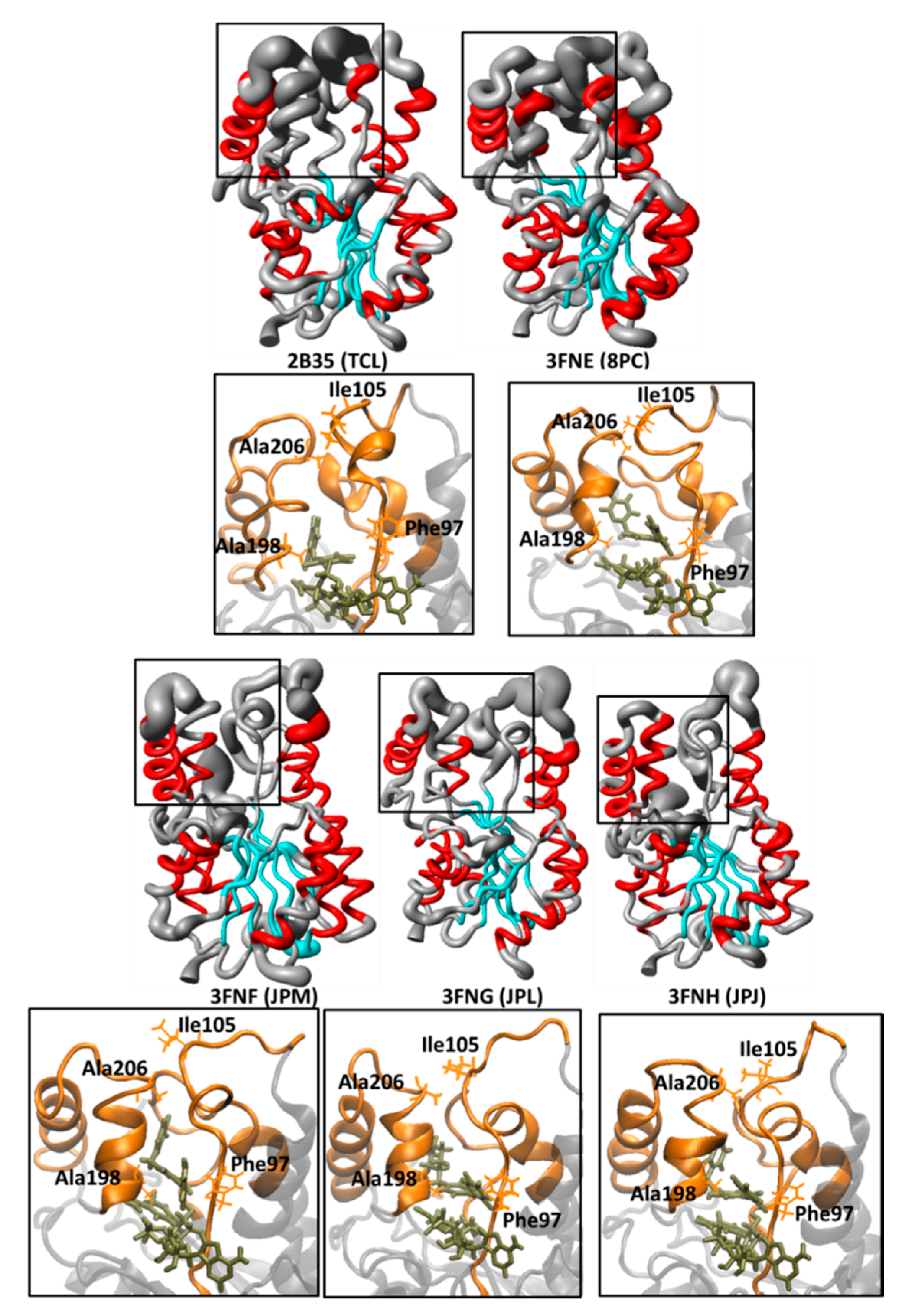
| 3FNF | 2.30 | NAD | JPM | 7.93 | [24] |
| 2B36 | 2.80 | NAD | 5PP | 7.91 | [23] |
| 3FNE | 1.98 | NAD | 8PC | 7.78 | [24] |
| 3FNH | 2.80 | NAD | JPJ | 7.52 | [24] |
| 4OXY | 2.35 | NAD | 1TN | 7.22 | [28] |
| 3FNG | 1.97 | NAD | JPL | 7.17 | [24] |
| 4OIM | 1.85 | NAD | JUS | 6.77 | [16] |
| 2B35 | 2.30 | NAD | TCL | 6.16 | [23] |
2. Results and Discussion
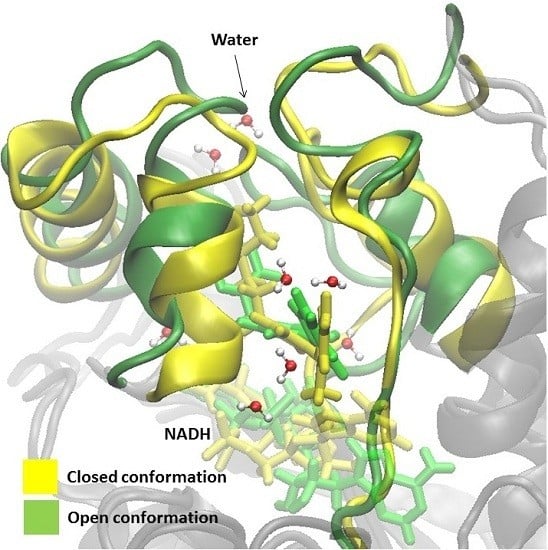
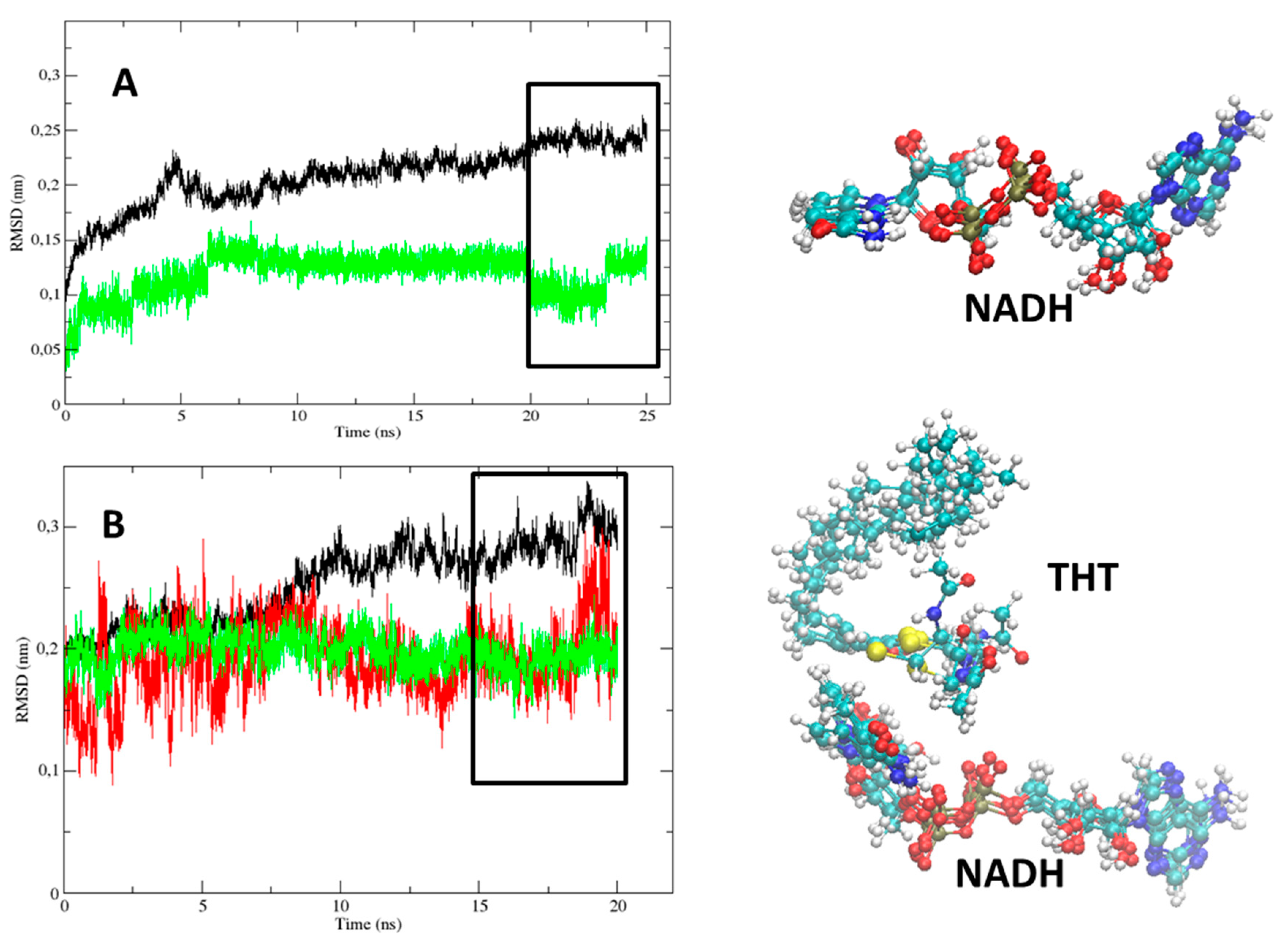
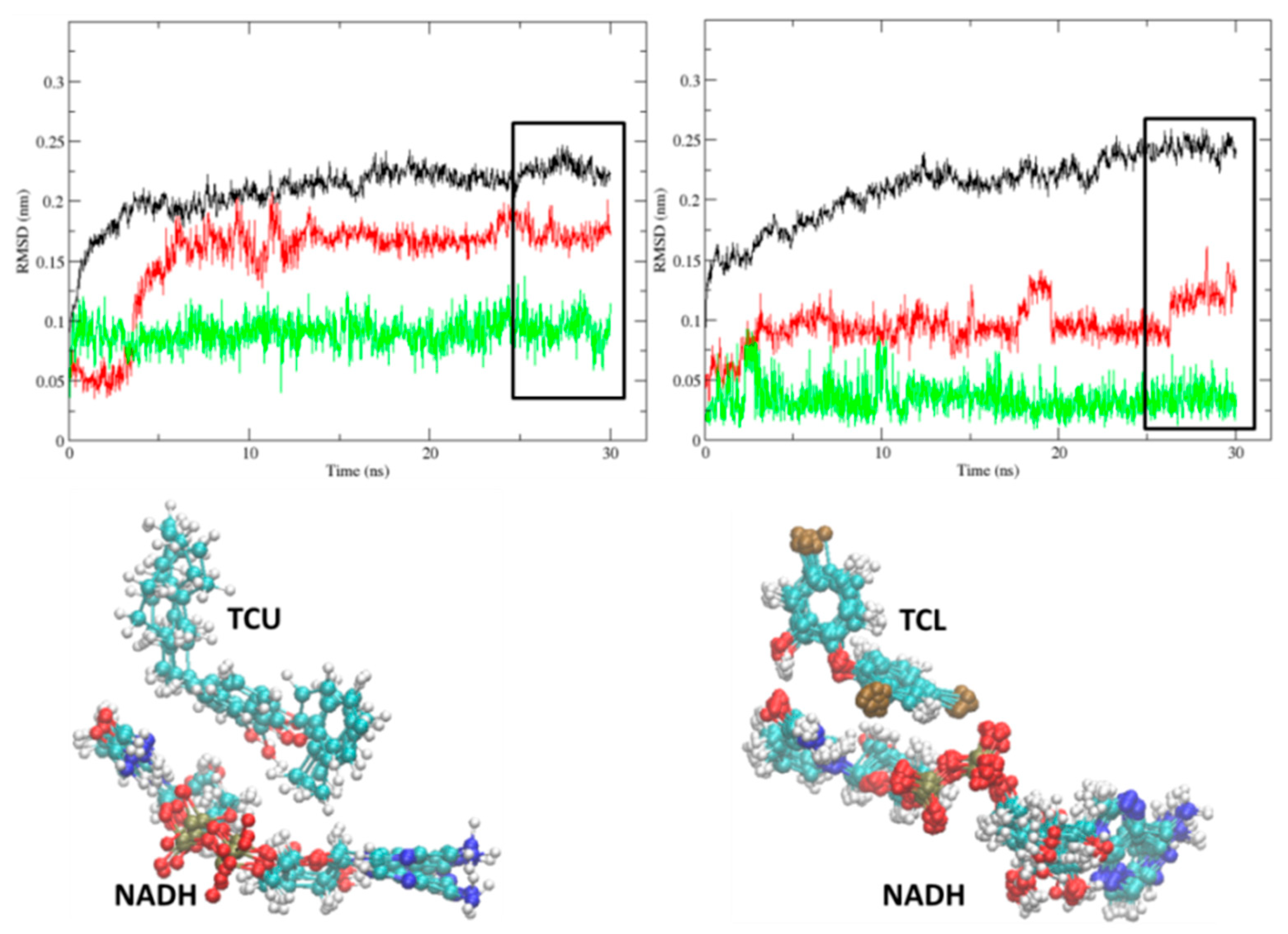
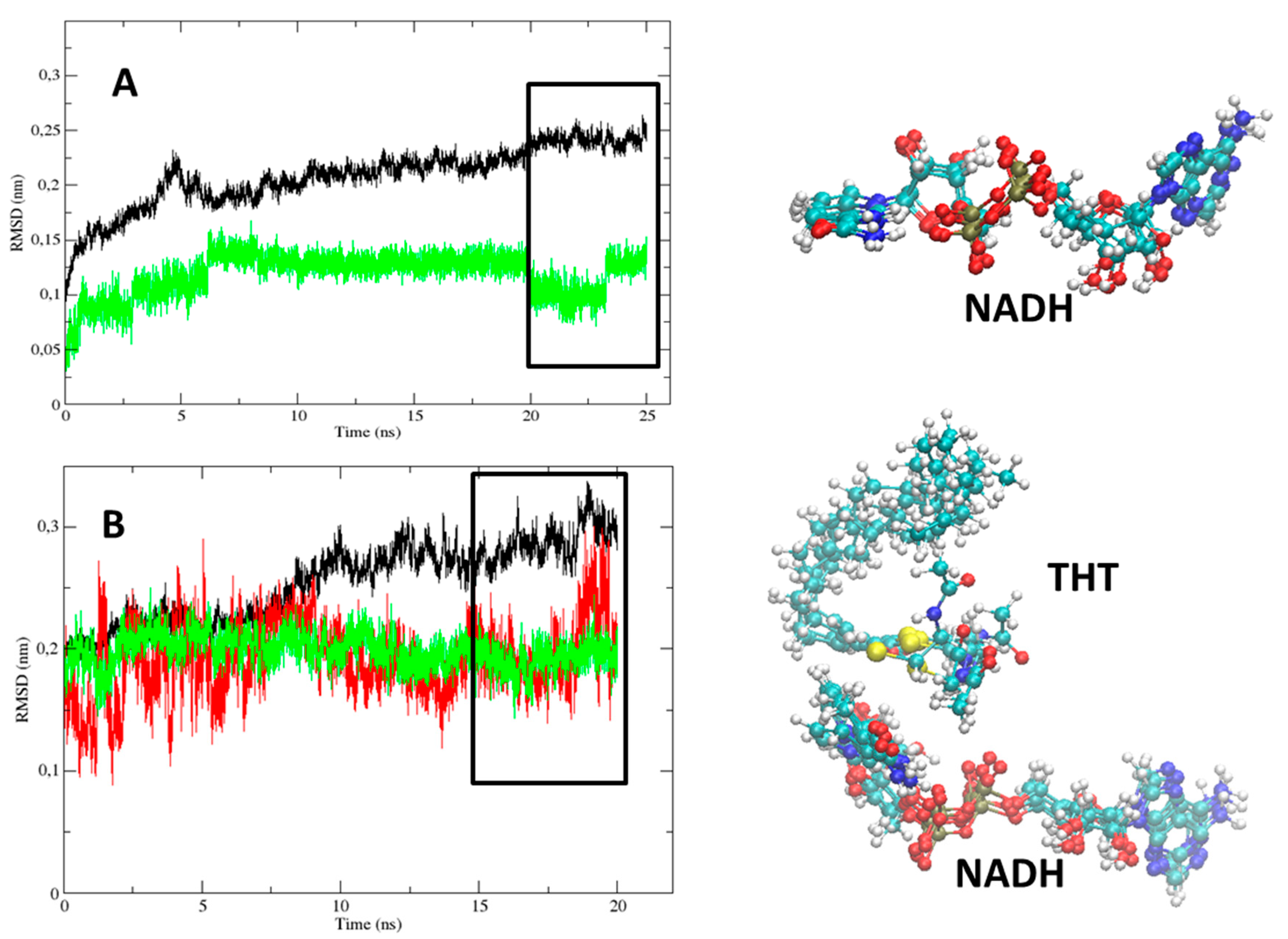
2.1. Comparative Analysis of the MD Simulations of the Holoenzyme (InhA-NADH) and Holoenzyme-Substrate (InhA-NADH-THT) Complexes


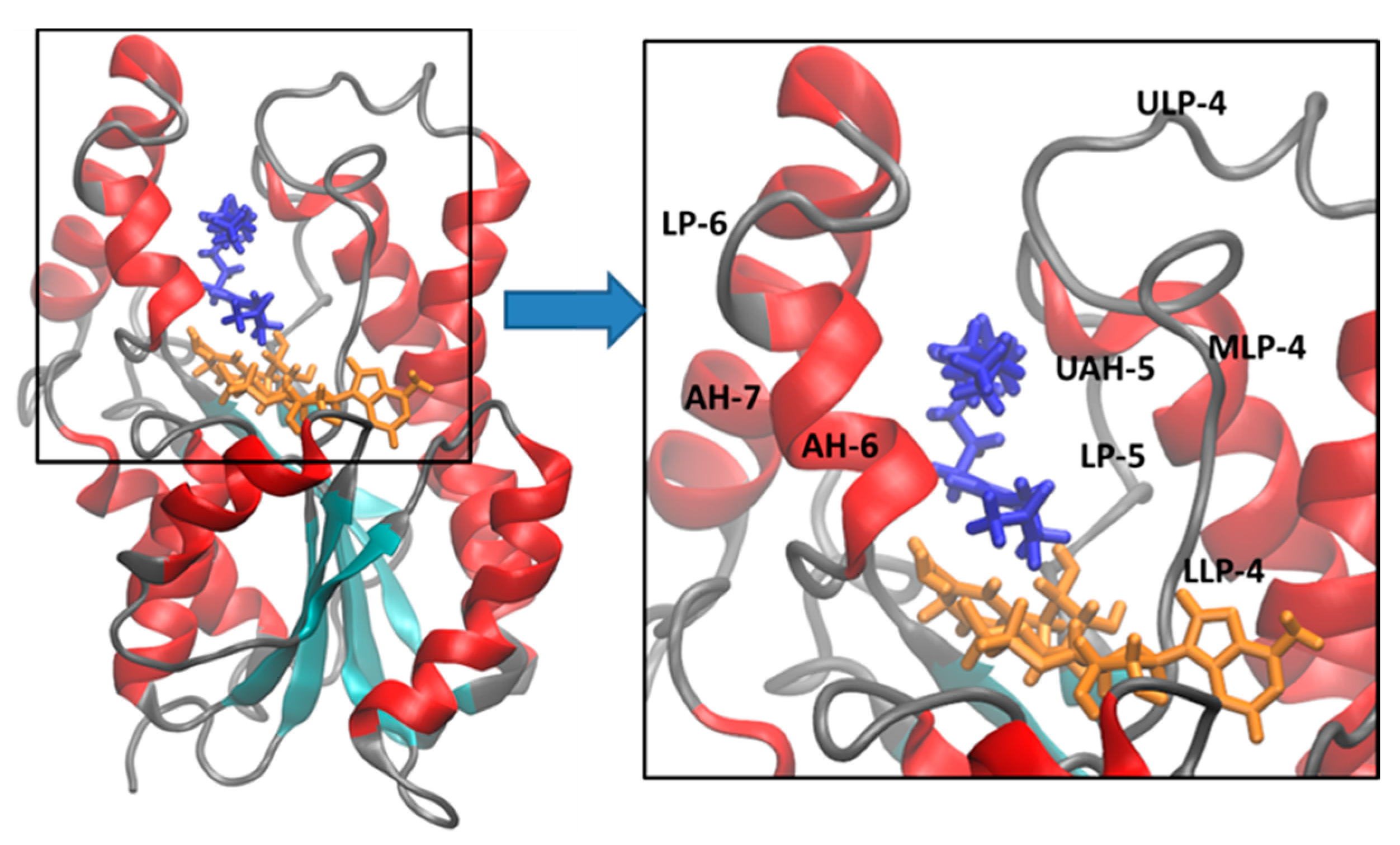
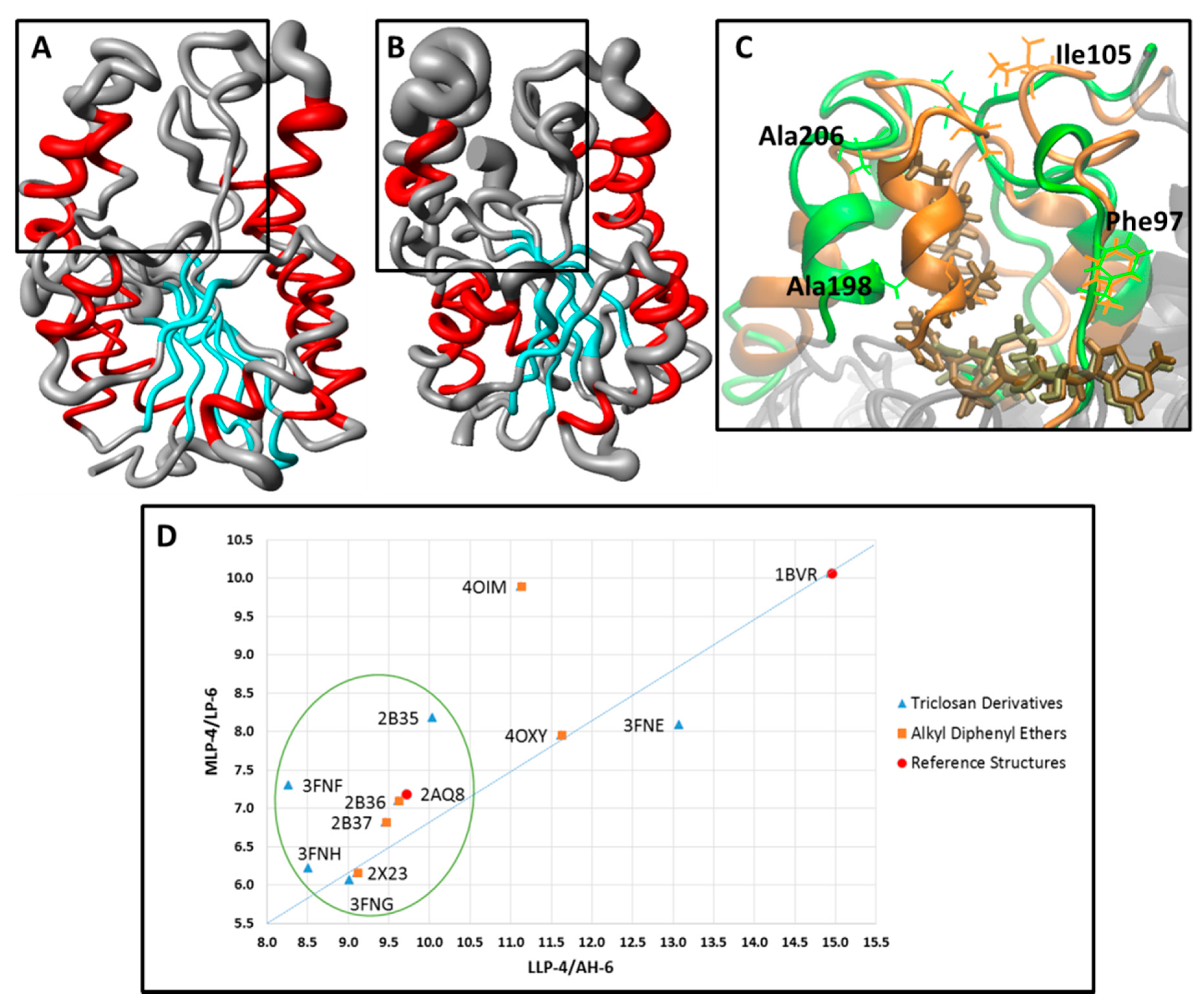
| # a | LLP-4/AH-6 b | MLP-4/LP-6 c | # a | LLP-4/AH-6 b | MLP-4/LP-6 c |
|---|---|---|---|---|---|
| 2AQ8 | 9.71 | 7.19 | 1BVR | 14.95 | 10.06 |
| 2X23 | 9.12 | 6.16 | 2B37 | 9.47 | 6.82 |
| 3FNF | 8.27 | 7.30 | 2B36 | 9.63 | 7.09 |
| 3FNE | 13.08 | 8.09 | 3FNH | 8.52 | 6.22 |
| 4OXY | 11.63 | 7.95 | 3FNG | 9.02 | 6.06 |
| 4OIM | 11.13 | 9.89 | 2B35 | 10.04 | 8.18 |
2.2. Comparative Analysis of the MD Simulations of the Holoenzyme-Inhibitor Complexes



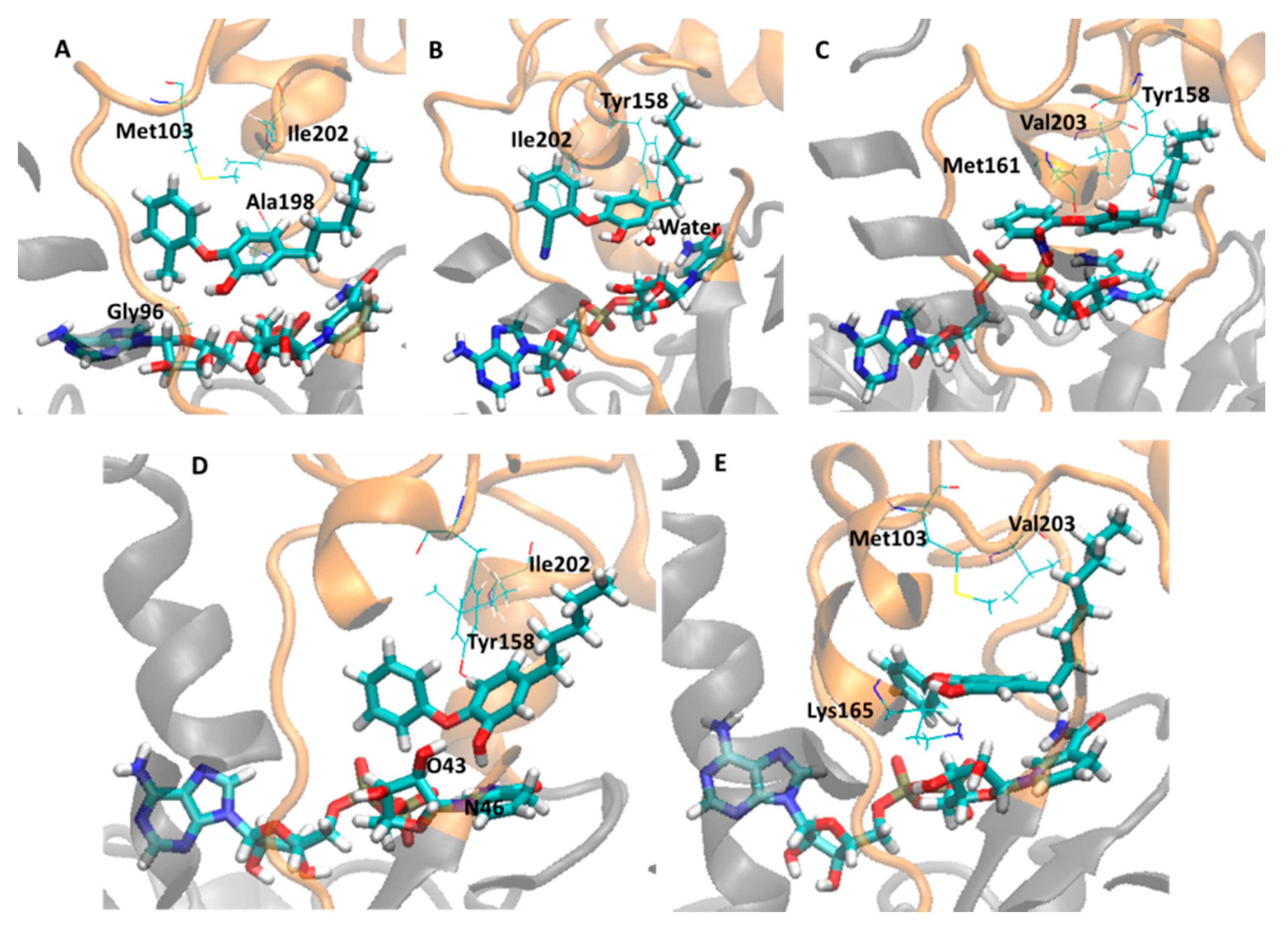
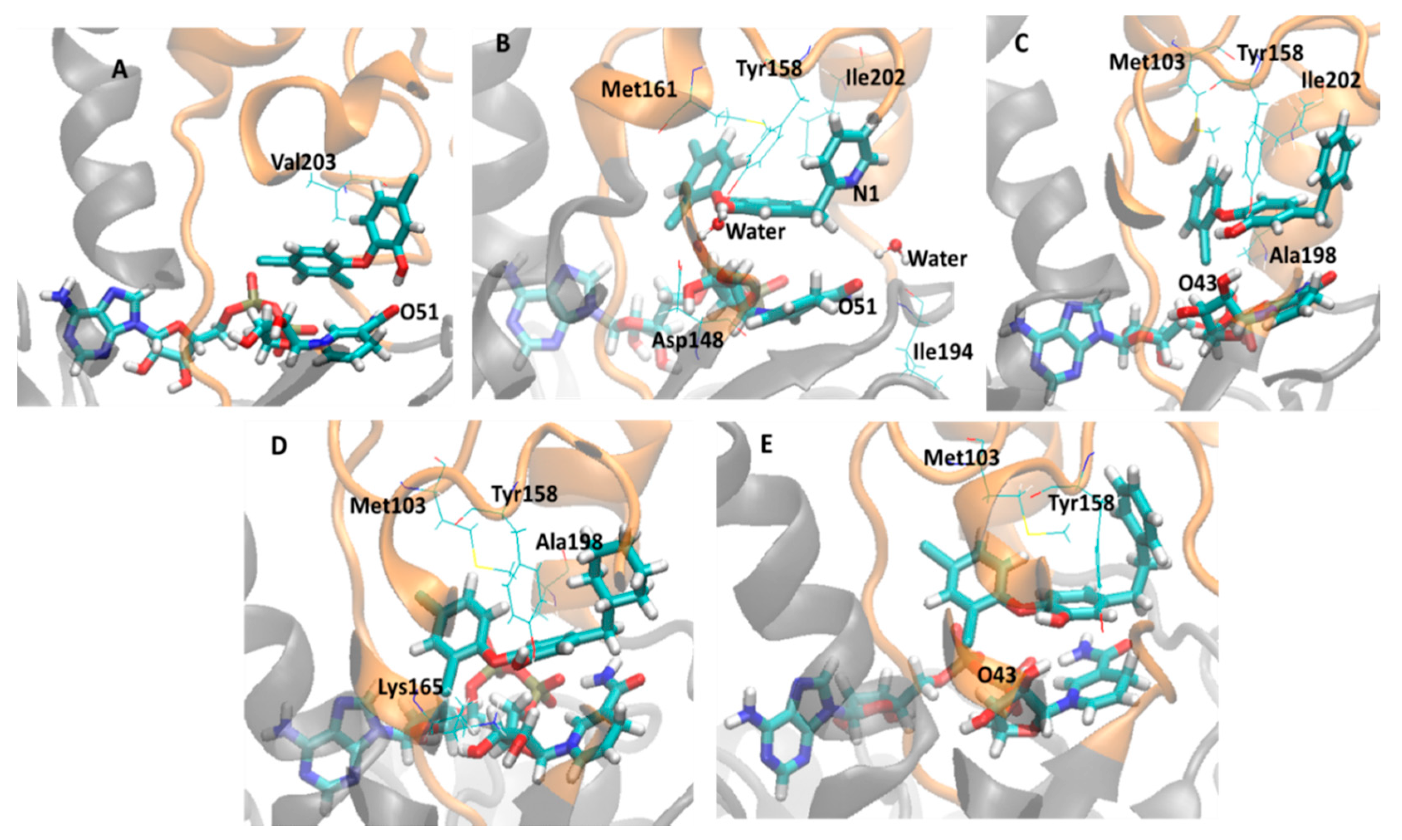
2.3. Analysis of Holoenzima-Inhibitor Interactions by Hydrogen Bond and Hydrophobic Contact

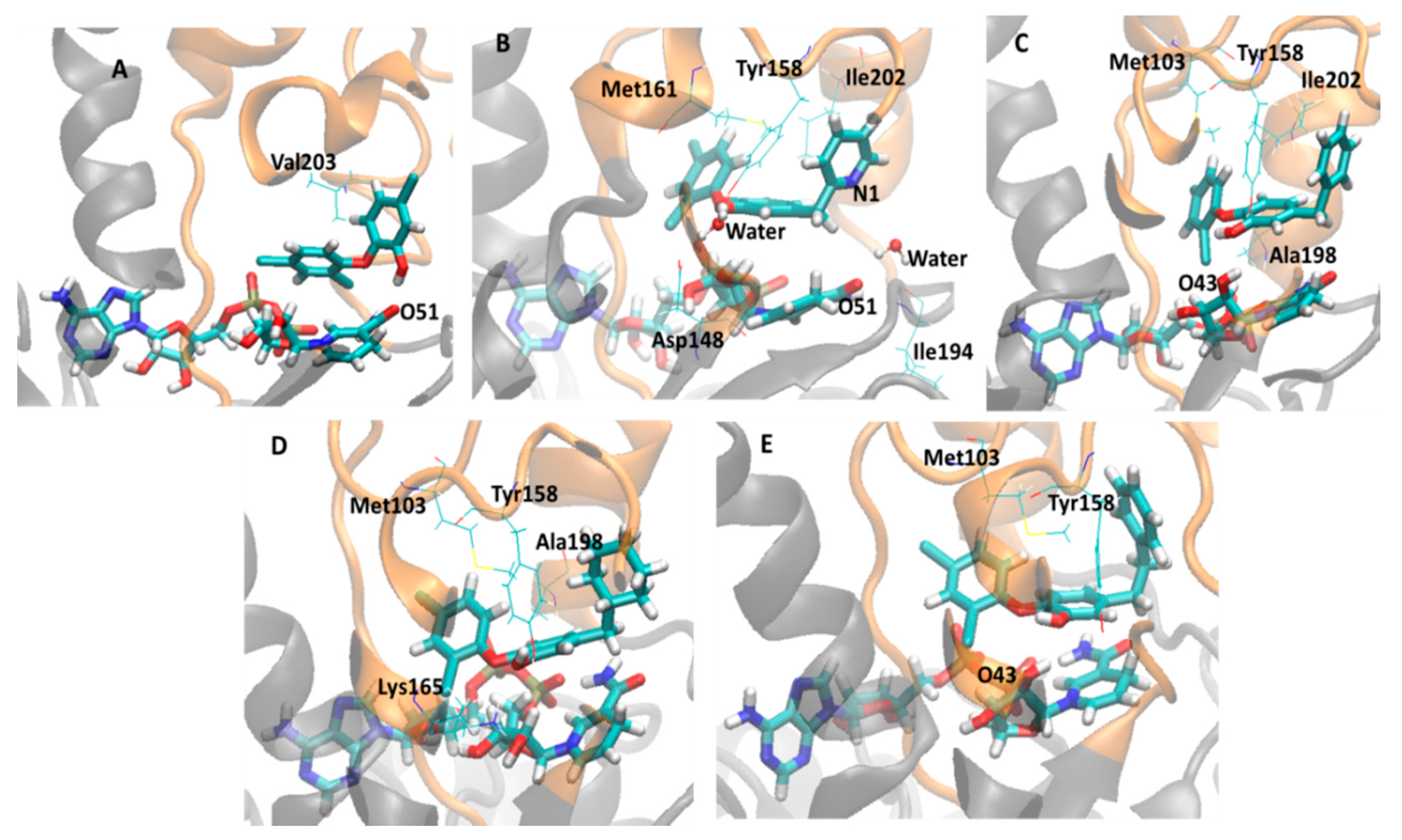
| # a | Hydrogen Bond b | Hydrophobic Contact c | ||||
|---|---|---|---|---|---|---|
| Donor | Acceptor | Time (%) d | Residue | Distance | Time (%) d | |
| 2X23 (TCU, 9.86) | OH | Gly96 | 26.5 | Met103 | 3.49 (± 0.26) | 92 |
| - | - | - | Ile202 | 3.80 (± 0.14) | 25.6 | |
| - | - | - | Ala198 | 3.83 (± 0.15) | 25.2 | |
| 2B37 (8PS, 8.47) | Lys165 | OH | 4.8 | Met103 | 3.91 (± 0.34) | 99 |
| - | - | - | Val203 | 3.57 (± 0.26) | 76.7 | |
| 3FNF (JPM, 7.93) | OH | Tyr158 | 42.0 | Met103 | 3.73 (± 0.19) | 65.7 |
| OH | NADH O43 | 27.2 | Ile202 | 3.82 (± 0.14) | 39.3 | |
| - | - | - | Ala198 | 3.82 (± 0.10) | 16.4 | |
| 2B36 (5PP, 7.91) | NADH O43 | OH | 87.6 | Tyr158 | 3.20 (± 0.25) | 95.4 |
| OH | NADH N46 | 28.3 | Ile202 | 3.88 (± 0.13) | 3.4 | |
| Tyr158 | OH | 5.5 | - | - | - | |
| 3FNE (8PC, 7.78) | OH | HOH | 98.5 | Met161 | 3.42 (± 0.32) | 92 |
| HOH | Tyr158 | 96.2 | Ile202 | 3.74 (± 0.18) | 30.5 | |
| HOH | Asp148 | 76.2 | - | - | - | |
| HOH | NADH O51 | 65.29 | - | - | - | |
| HOH | N1 | 57.7 | - | - | - | |
| HOH | Ile194 | 38 | - | - | - | |
| 3FNH (JPJ, 7.52) | OH | NADH O43 | 25.0 | Met103 | 2.88 (± 0.25) | 100 |
| OH | Tyr158 | 23.7 | - | - | - | |
| 4OXY (1TN, 7.22) | Tyr158 | OH | 91.4 | Met161 | 3.70 (± 0.19) | 82.4 |
| - | - | - | Val203 | 3.71 (± 0.19) | 37 | |
| 3FNG (JPL, 7.17) | OH | Tyr158 | 73.6 | Met103 | 3.30 (± 0.32) | 94.4 |
| Lys165 | OH | 67 | Ala198 | 3.88 (± 0.10) | 4.0 | |
| 4OIM (JUS, 6.77) | HOH | Tyr158 | 98.2 | Tyr158 | 3.72 (± 0.18) | 71.1 |
| HOH | OH | 89.2 | Ile202 | 3.94 (± 0.07) | 2.2 | |
| 2B35 (TCL, 6.16) | OH | NADH O51 | 96.8 | Val203 | 3.81 (± 0.14) | 40.7 |
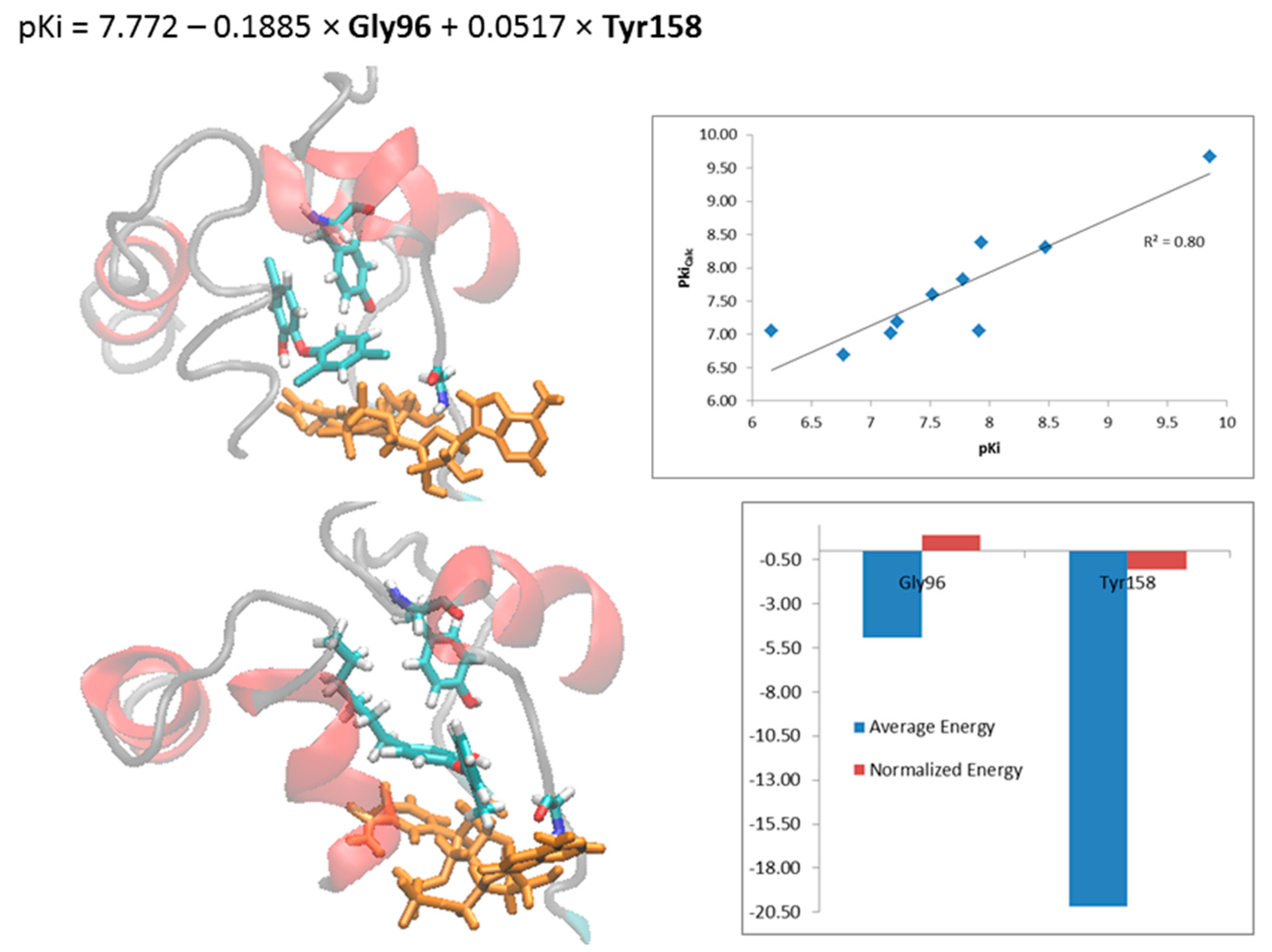
2.4. Steric and Electrostatic Interactions of the Holoenzyme-Inhibitor Complexes

2.5. Final Considerations
3. Experimental Section
3.1. Structure of Protein Complexes Selection
3.2. Biological Data
3.3. Molecular Docking Simulation
3.4. Molecular Dynamics Simulations
3.5. Molecular Dynamics Analysis
3.6. Steric and Electrostatic Interactions Calculations of the Holoenzyme-Inhibitor Complexes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carrol, E.D.; Clark, J.E.; Cant, A.J. Non-pulmonary tuberculosis. Paediatr. Respir. Rev. 2001, 2, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, S.; Nair, P.P.; Emmanuel, R.V; Agrawal, K. Diversities in presentations of extrapulmonary tuberculosis. BMJ Case Rep. 2013, 2013, 1–5. [Google Scholar]
- Golden, M.P.; Vikram, H.R. Extrapulmonary tuberculosis: An overview. Am. Fam. Physician 2005, 72, 1761–1768. [Google Scholar] [PubMed]
- WHO. Global Tuberculosis Report 2014; Technical Report; WHO Press: Geneva, Switzerland, 2014. [Google Scholar]
- Dye, C.; Watt, C.J.; Bleed, D.M.; Hosseini, S.M.; Raviglione, M.C. Evolution of tuberculosis control and prospects for reducing tuberculosis incidence, prevalence, and deaths globally. J. Am. Med. Assoic. 2005, 293, 2767–2775. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.C.; Whalen, C.C. Non-clinical factors associated with TB: Important for DOTS impact evaluation and disease elimination. Trans. R. Soc. Trop. Med. Hyg. 2014, 108, 523–525. [Google Scholar] [CrossRef] [PubMed]
- De Souza, M.V.N. Promising drugs against tuberculosis. Recent Pat. Antiinfect. Drug Discov. 2006, 1, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Raviglione, M.; Hafner, R.; Fordham von Reyn, C. Tuberculosis. N. Engl. J. Med. 2013, 368, 745–755. [Google Scholar] [CrossRef] [PubMed]
- De Souza, M.V.N. Promising candidates in clinical trials against multidrug-resistant tuberculosis (MDR-TB) based on natural products. Fitoterapia 2009, 80, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Massengo-Tiassé, R.P.; Cronan, J.E. Diversity in enoyl-acyl carrier protein reductases. Cell. Mol. Life Sci. 2009, 66, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Beld, J.; Lee, D.J.; Burkart, M.D. Fatty acid biosynthesis revisited: Structure elucidation and metabolic engineering. Mol. Biosyst. 2015, 11, 38–59. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.C.; Chen, Y.M.A.; Chen, H.Y.; Jou, R. Single nucleotide polymorphisms in cell wall biosynthesis-associated genes and phylogeny of Mycobacterium tuberculosis lineages. Infect. Genet. Evol. 2010, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.V.; Reich, R.; Dou, S.J.; Jasperse, L.; Pan, X.; Wanger, A.; Quitugua, T.; Graviss, E.A. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Feuerriegel, S.; Köser, C.U.; Niemann, S. Phylogenetic polymorphisms in antibiotic resistance genes of the Mycobacterium tuberculosis complex. J. Antimicrob. Chemother. 2014, 69, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, T.; Koseki, Y.; Kobayashi, M.; Yamada, A.; Morita, K.; Yamaguchi, K.; Tsurusawa, R.; Gulten, G.; Komatsu, H.; Sakamoto, H.; et al. Identification of compounds with potential antibacterial activity against Mycobacterium through structure-based drug screening. J. Chem. Inf. Model. 2013, 53, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Knudson, S.E.; Bommineni, G.R.; Li, H.J.; Lai, C.T.; Liu, N.; Garcia-Diaz, M.; Simmerling, C.; Patil, S.S.; Slayden, R.A.; Tonge, P.J. Time-dependent diaryl ether inhibitors of InhA: Structure-activity relationship studies of enzyme inhibition, antibacterial activity, and in vivo efficacy. ChemMedChem 2014, 9, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Kannan, S.; Wichapong, K.; Owono Owono, L.C.; Sippl, W.; Megnassan, E. Binding of pyrazole-based inhibitors to Mycobacterium tuberculosis pantothenate synthetase: Docking and MM-GB(PB)SA analysis. Mol. Biosyst. 2014, 10, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Mohan Krishna, K.; Inturi, B.; Pujar, G.V.; Purohit, M.N.; Vijaykumar, G.S. Design, synthesis and 3D-QSAR studies of new diphenylamine containing 1,2,4-triazoles as potential antitubercular agents. Eur. J. Med. Chem. 2014, 84, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Yazdankhah, S.P.; Scheie, A.A.; Høiby, E.A.; Lunestad, B.-T.; Heir, E.; Fotland, T.Ø.; Naterstad, K.; Kruse, H. Triclosan and antimicrobial resistance in bacteria: An overview. Microb. Drug Resist. 2006, 12, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Yan, K.; Wallis, N.G.; Reed, S.; Moore, T.D.; Rittenhouse, S.F.; DeWolf, W.E.; Huang, J.; McDevitt, D.; Miller, W.H.; et al. Defining and combating the mechanisms of triclosan resistance in clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 2002, 46, 3343–3347. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Dixit, S.; More, U.; Aminabhavi, T.; Kulkarni, V.; Gadad, A. Enoyl ACP reductase as effective target for the synthesized novel antitubercular drugs: A-State-of-the-Art. MiniRev. Med. Chem. 2014, 14, 678–693. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.J.; Truglio, J.J.; Boyne, M.E.; Novichenok, P.; Zhang, X.; Stratton, C.F.; Li, H.J.; Kaur, T.; Amin, A.; Johnson, F.; Slayden, R.A.; et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Freundlich, J.S.; Wang, F.; Vilchèze, C.; Gulten, G.; Langley, R.; Schiehser, G.A.; Jacobus, D.P.; Jacobs, W.R.; Sacchettini, J.C. Triclosan derivatives: Towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. ChemMedChem 2009, 4, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Luckner, S.R.; Liu, N.; am Ende, C.W.; Tonge, P.J.; Kisker, C. A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 2010, 285, 14330–14337. [Google Scholar] [CrossRef] [PubMed]
- Roujeinikova, A.; Levy, C.W.; Rowsell, S.; Sedelnikova, S.; Baker, P.J.; Minshull, C.A.; Mistry, A.; Colls, J.G.; Camble, R.; Stuitje, A.R.; et al. Crystallographic analysis of triclosan bound to enoyl reductase. J. Mol. Biol. 1999, 294, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.L.; Xiao, G.; Tonge, P.J. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry 2000, 39, 7645–7650. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Lai, C.T.; Pan, P.; Yu, W.; Liu, N.; Bommineni, G.R.; Garcia-Diaz, M.; Simmerling, C.; Tonge, P.J. A structural and energetic model for the slow-onset inhibition of the mycobacterium tuberculosis ENoyl-ACP reductase InhA. ACS Chem. Biol. 2014, 9, 986–993. [Google Scholar] [CrossRef] [PubMed]
- am Ende, C.W.; Knudson, S.E.; Liu, N.; Childs, J.; Sullivan, T.J.; Boyne, M.; Xu, H.; Gegina, Y.; Knudson, D.L.; Johnson, F.; et al. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorgan. Med. Chem. Lett. 2008, 18, 3029–3033. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sobhia, M.E. Insights into the bonding pattern for characterizing the open and closed state of the substrate-binding loop in Mycobacterium tuberculosis InhA. Future Med. Chem. 2014, 6, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sobhia, M.E. Characterisation of the flexibility of substrate binding loop in the binding of direct InhA inhibitors. Int. J. Comput. Biol. Drug Des. 2013, 6, 318–342. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Madhavapeddi, P.; Naik, M.; Murugan, K.; Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate, G.; et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.H.; Rao, S.P.; Kondreddi, R.R.; Noble, C.G.; Camacho, L.R.; Tan, B.H.; Ng, S.H.; Ng, P.S.; Ma, N.L.; Lakshminarayana, S.B.; et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, P.S.M.; Ducati, R.G.; Basso, L.A.; Santos, D.S.; da Silva, L.H.P. Enzyme mechanism and slow-onset inhibition of plasmodium falciparum Enoyl-Acyl carrier protein reductase by an inorganic complex. Enzyme Res. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.L.P.; Pauli, I.; Dorn, M.; Schroeder, E.K.; Zhan, C.-G.; de Souza, O.N. Conformational changes in 2-trans-enoyl-ACP (CoA) reductase (InhA) from M. tuberculosis induced by an inorganic complex: A molecular dynamics simulation study. J. Mol. Model. 2012, 18, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.S.; Pereira, J.H.; Canduri, F.; Rodrigues, N.C.; de Souza, O.N.; de Azevedo, W.F.; Basso, L.A; Santos, D.S. Crystallographic and pre-steady-state kinetics studies on binding of NADH to wild-type and isoniazid-resistant enoyl-ACP(CoA) reductase enzymes from Mycobacterium tuberculosis. J. Mol. Biol. 2006, 359, 646–666. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Schiebel, J.; Yu, W.; Bommineni, G.R.; Pan, P.; Baxter, M.V.; Khanna, A.; Sotriffer, C.A.; Kisker, C.; Tonge, P.J. Rational optimization of drug-target residence time: Insights from inhibitor binding to the staphylococcus aureus FabI enzyme-product complex. Biochemistry 2013, 52, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.J.; Parikh, S.; Xiao, G.; Tonge, P.J.; Kisker, C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 1999, 290, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Mehboob, S.; Truong, K.; Santarsiero, B.D.; Johnson, M.E. Structure of the Francisella tularensis enoyl-acyl carrier protein reductase (FabI) in complex with NAD+ and triclosan. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-H.; Park, J.; Kim, E.; Hohng, S.; Kim, H.-S. Protein conformational dynamics dictate the binding affinity for a ligand. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.J.; Mauldin, R. V; Gromova, A. V; Singleton, S.F.; Collins, E.J.; Lee, A.L. Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat. Chem. Biol. 2012, 8, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Fatmi, M.Q.; Ai, R.; Chang, C.E.A. Synergistic regulation and ligand-induced conformational changes of tryptophan synthase. Biochemistry 2009, 48, 9921–9931. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Gerstein, M. Relating protein conformational changes to packing efficiency and disorder. Protein Sci. 2009, 18, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Kamsri, P.; Koohatammakun, N.; Srisupan, A; Meewong, P.; Punkvang, A; Saparpakorn, P.; Hannongbua, S.; Wolschann, P.; Prueksaaroon, S.; Leartsakulpanich, U.; et al. Rational design of InhA inhibitors in the class of diphenyl ether derivatives as potential anti-tubercular agents using molecular dynamics simulations. SAR QSAR Environ. Res. 2014, 25, 473–488. [Google Scholar] [PubMed]
- Kamsri, P.; Punkvang, A.; Saparpakorn, P.; Hannongbua, S.; Irle, S.; Pungpo, P. Elucidating the structural basis of diphenyl ether derivatives as highly potent enoyl-ACP reductase inhibitors through molecular dynamics simulations and 3D-QSAR study. J. Mol. Model. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Ward, W.H.J.; Holdgate, G.A.; Rowsell, S.; McLean, E.G.; Pauptit, R.A.; Clayton, E.; Nichols, W.W.; Colls, J.G.; Minshull, C.A.; Jude, D.A.; et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry 1999, 38, 12514–12525. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.L.; Xiao, G.; Tonge, P.J. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry 2000, 39, 7645–7650. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hopfinger, A J. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J. Chem. Inf. Model. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Vilchèze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, L.; Wang, X.; Wang, X.; Liu, H.; Qian, X.; Zhu, Y.; Yu, H. Molecular docking, molecular dynamics simulation, and structure-based 3D-QSAR studies on estrogenic activity of hydroxylated polychlorinated biphenyls. Sci. Total Environ. 2012, 441, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.Q.; de Brito, M.A.; Hoelz, L.V.B.; de Alencastro, R.B.; Castro, H.C.; Rodrigues, C.R.; Albuquerque, M.G. Receptor-dependent (RD) 3D-QSAR approach of a series of benzylpiperidine inhibitors of human acetylcholinesterase (HuAChE). Eur. J. Med. Chem. 2011, 46, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.Q.; Lima, J.A.; Pinto, A.D.C.; de Alencastro, R.B.; Albuquerque, M.G. Docking of the alkaloid geissospermine into acetylcholinesterase: A natural scaffold targeting the treatment of Alzheimer’s disease. J. Mol. Model. 2011, 17, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Onodera, K.; Satou, K.; Hirota, H. Evaluations of molecular docking programs for virtual screening. J. Chem. Inf. Model. 2007, 47, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Brooijmans, N.; Kuntz, I.D. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Łaźniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDB bind database. J. Comput. Chem. 2011, 32, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: Comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; Hess, B.; Lindahl, E. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Da Silva, S.; Alan, W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser interfacE. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.A.S.T.; Horta, B.A.C.; De Alencastro, R.B. MKTOP: A program for automatic construction of molecular topologies. J. Braz. Chem. Soc. 2008, 19, 1433–1435. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Kusalik, P.G.; Svishchev, I.M. The spatial structure in liquid water. Science 1994, 265, 1219–1221. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- HbMap2Grace, version 1.0; Software for mapping the hydrogen bond frequency; LMDM: Rio de Janeiro, Brazil, 2009.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grace, version 5.1.23; 2D plotting tool; Plasma Laboratory—Weizmann Institute of Science: Rehovot, Israel, 1996.
- Koradi, R.; Billeter, M.; Wüthrich, K. Molmol: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, C.H.d.S.; De Alencastro, R.B.; Kaiser, C.R.; De Souza, M.V.N.; Rodrigues, C.R.; Albuquerque, M.G. Aqueous Molecular Dynamics Simulations of the M. tuberculosis Enoyl-ACP Reductase-NADH System and Its Complex with a Substrate Mimic or Diphenyl Ethers Inhibitors. Int. J. Mol. Sci. 2015, 16, 23695-23722. https://doi.org/10.3390/ijms161023695
Lima CHdS, De Alencastro RB, Kaiser CR, De Souza MVN, Rodrigues CR, Albuquerque MG. Aqueous Molecular Dynamics Simulations of the M. tuberculosis Enoyl-ACP Reductase-NADH System and Its Complex with a Substrate Mimic or Diphenyl Ethers Inhibitors. International Journal of Molecular Sciences. 2015; 16(10):23695-23722. https://doi.org/10.3390/ijms161023695
Chicago/Turabian StyleLima, Camilo Henrique da Silva, Ricardo Bicca De Alencastro, Carlos Roland Kaiser, Marcus Vinícius Nora De Souza, Carlos Rangel Rodrigues, and Magaly Girão Albuquerque. 2015. "Aqueous Molecular Dynamics Simulations of the M. tuberculosis Enoyl-ACP Reductase-NADH System and Its Complex with a Substrate Mimic or Diphenyl Ethers Inhibitors" International Journal of Molecular Sciences 16, no. 10: 23695-23722. https://doi.org/10.3390/ijms161023695