
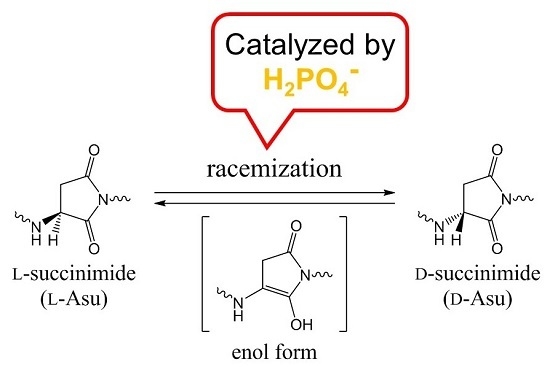
Racemization of the Succinimide Intermediate Formed in Proteins and Peptides: A Computational Study of the Mechanism Catalyzed by Dihydrogen Phosphate Ion


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
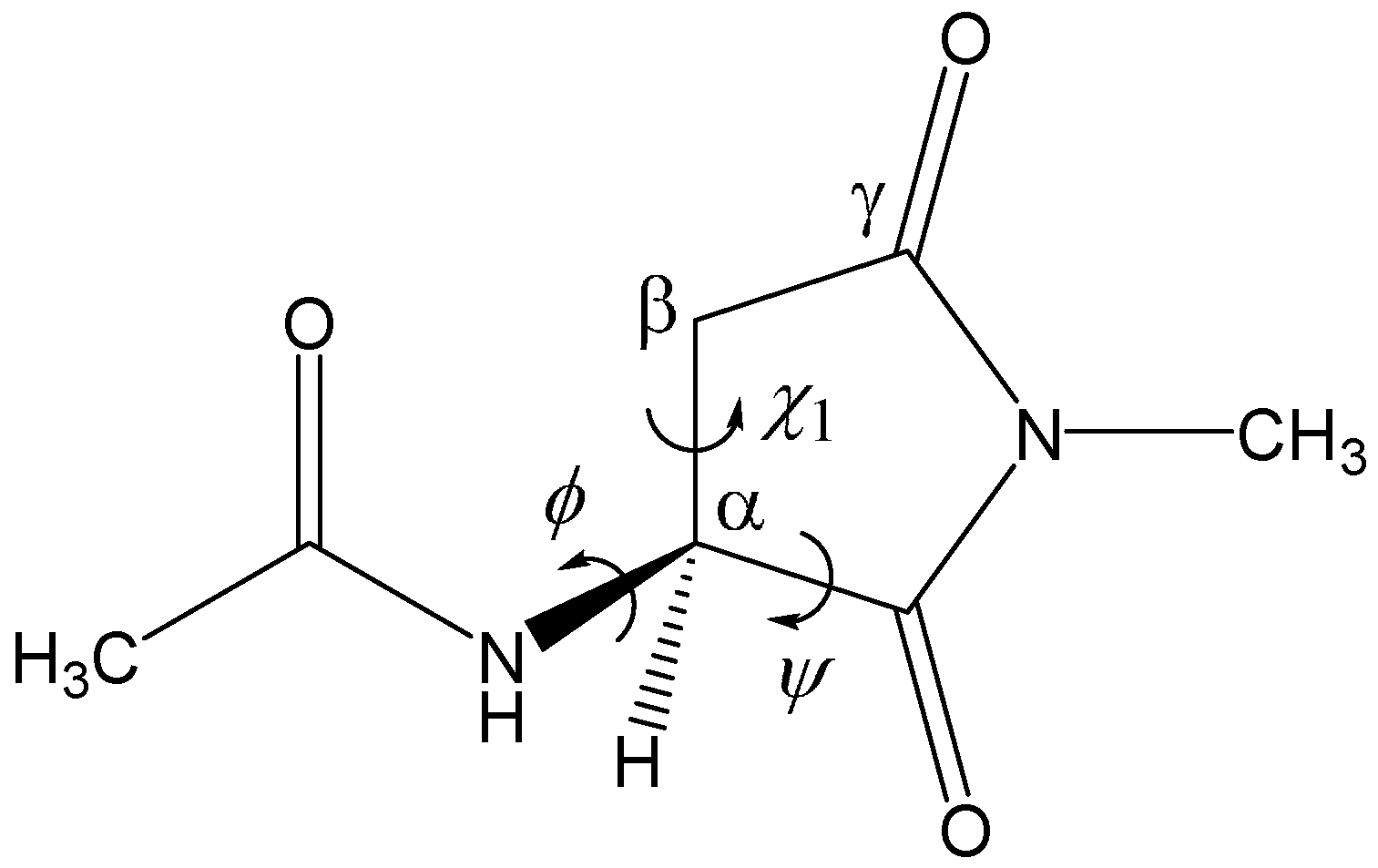
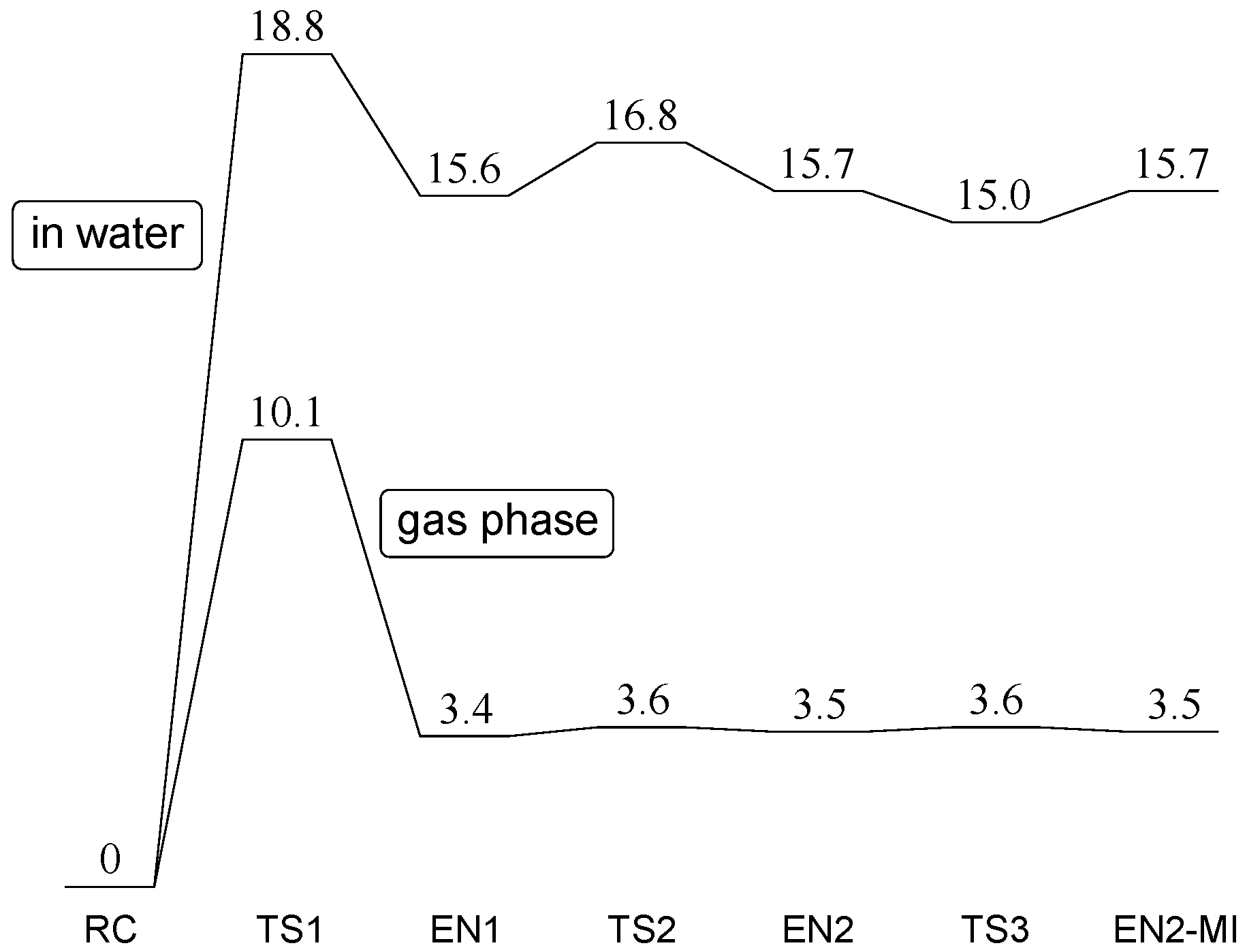
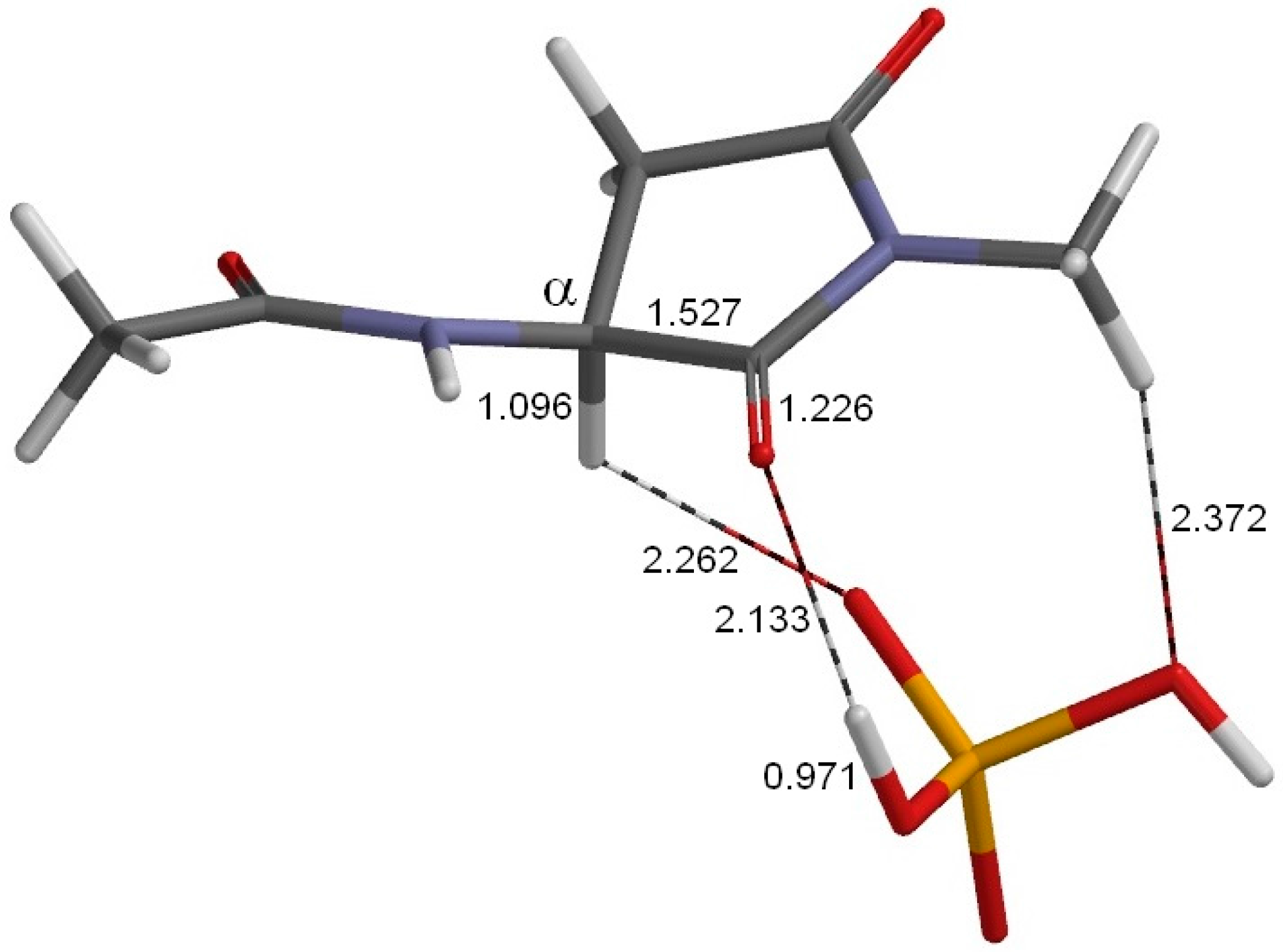
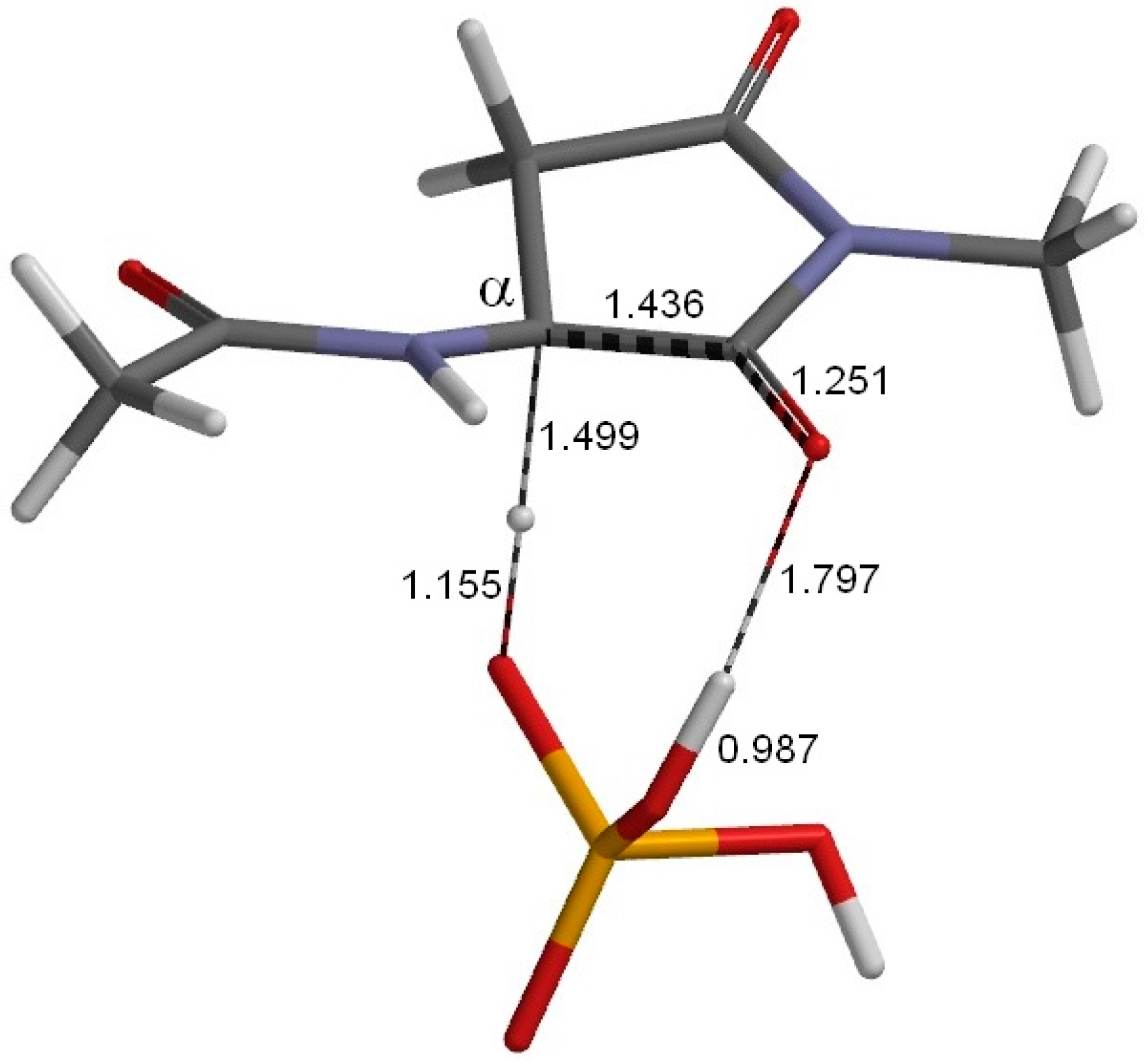
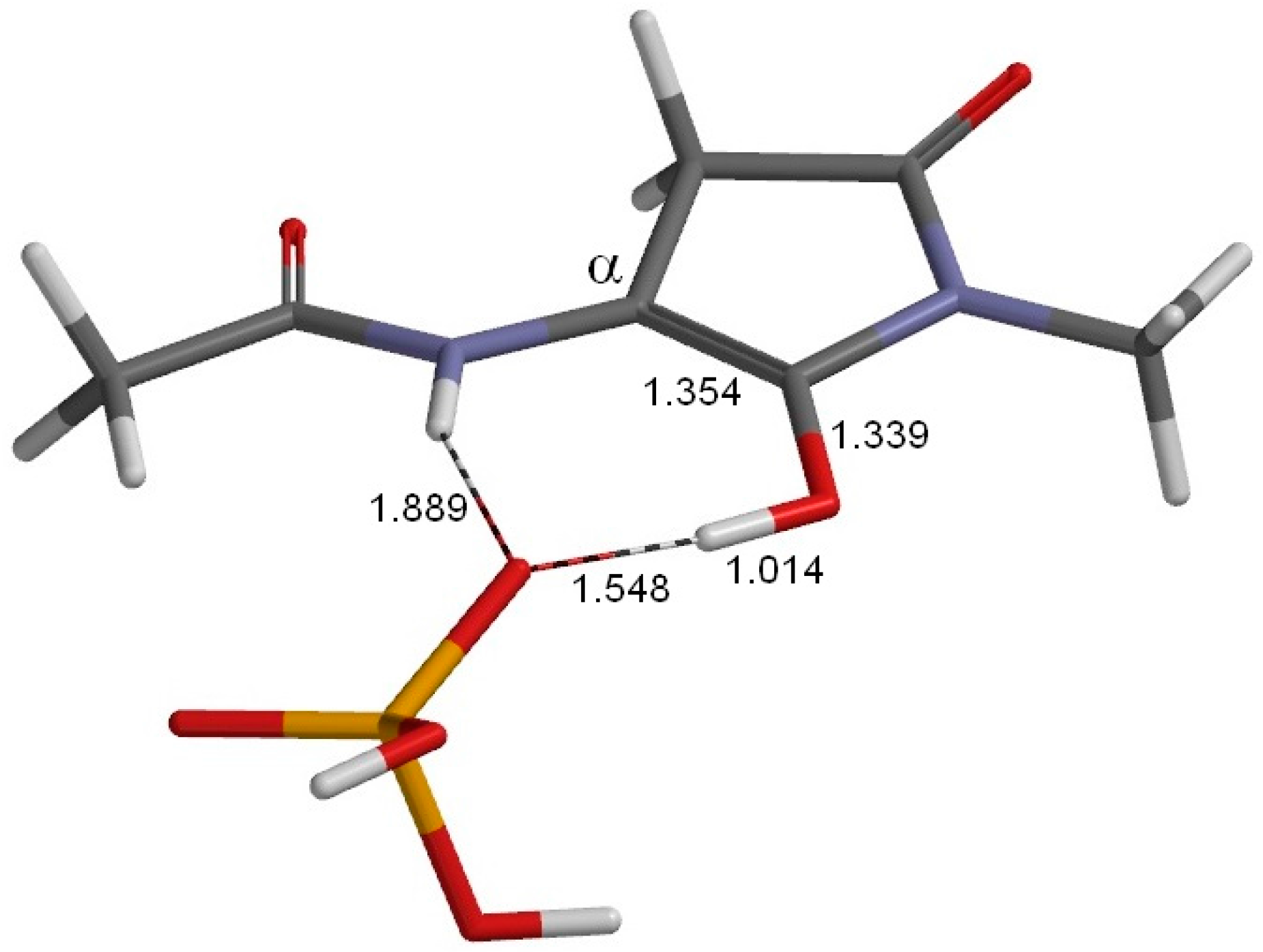
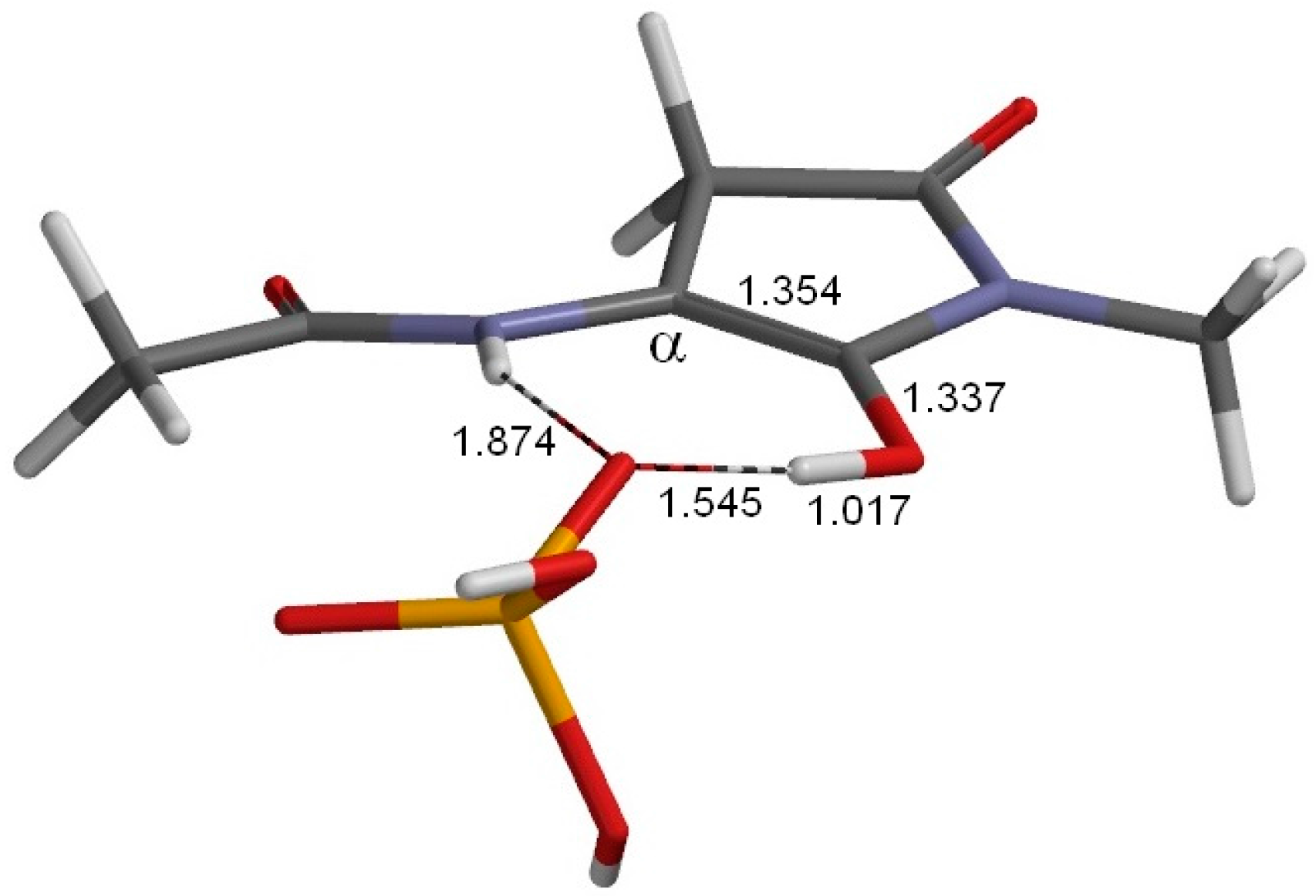
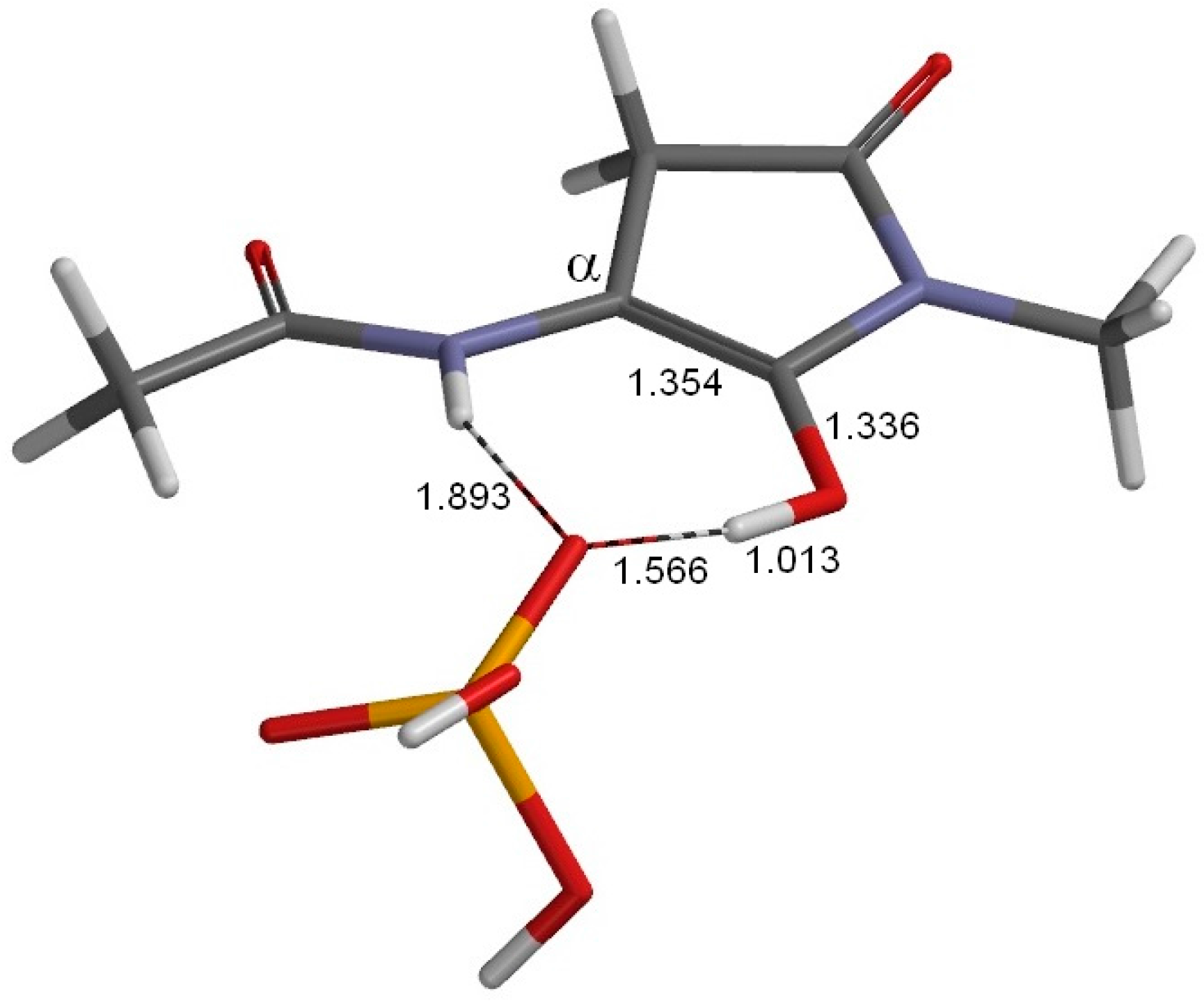
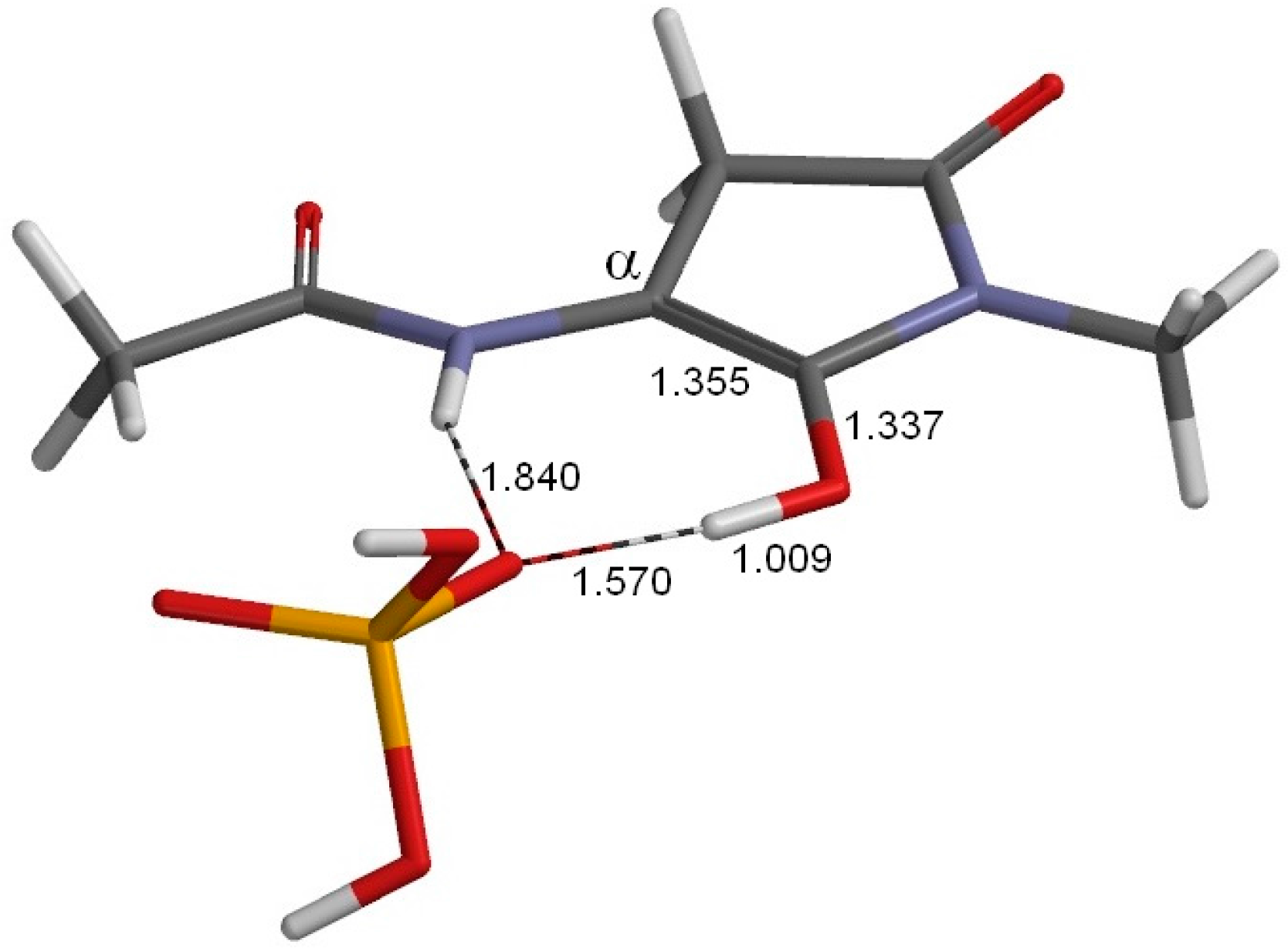
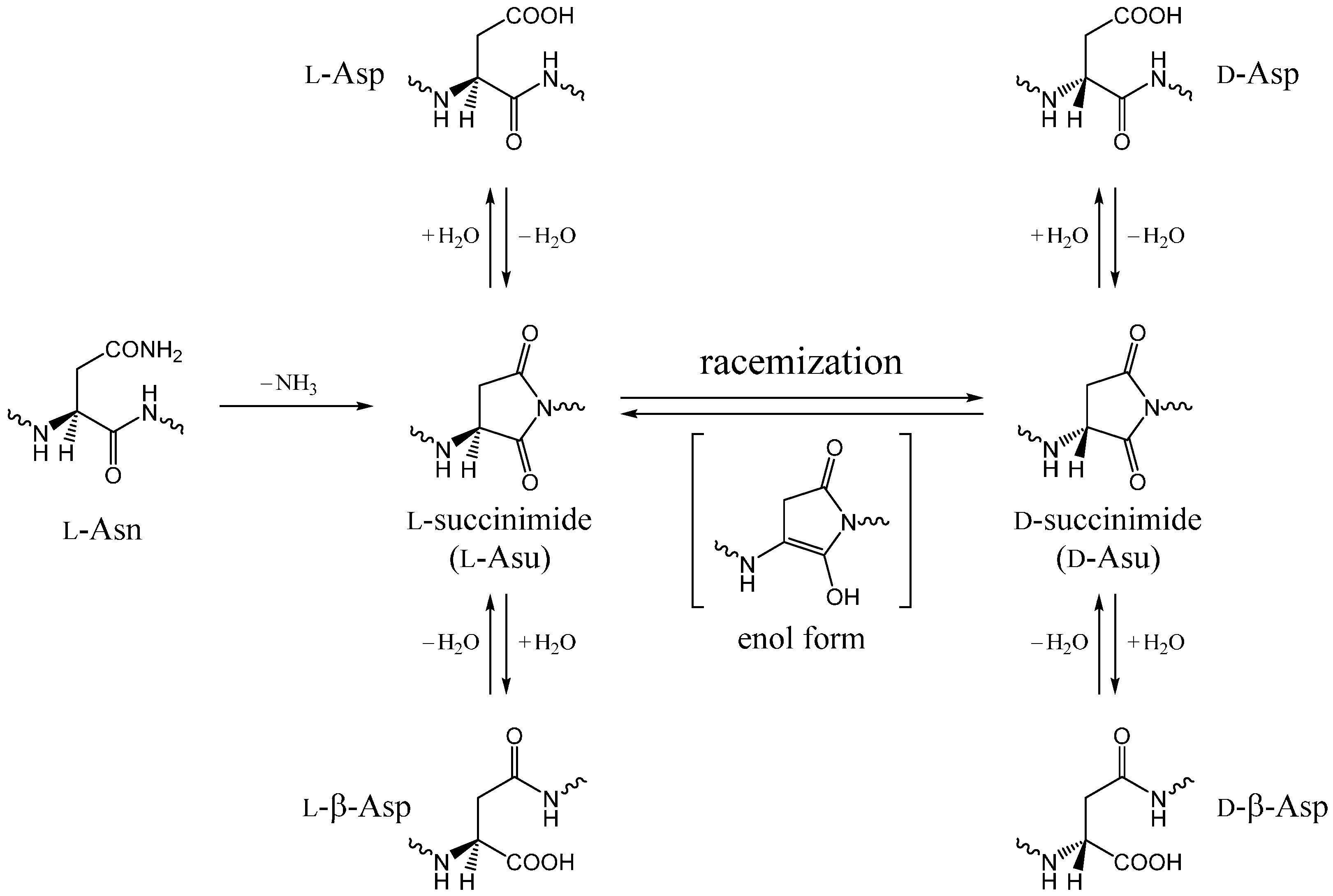
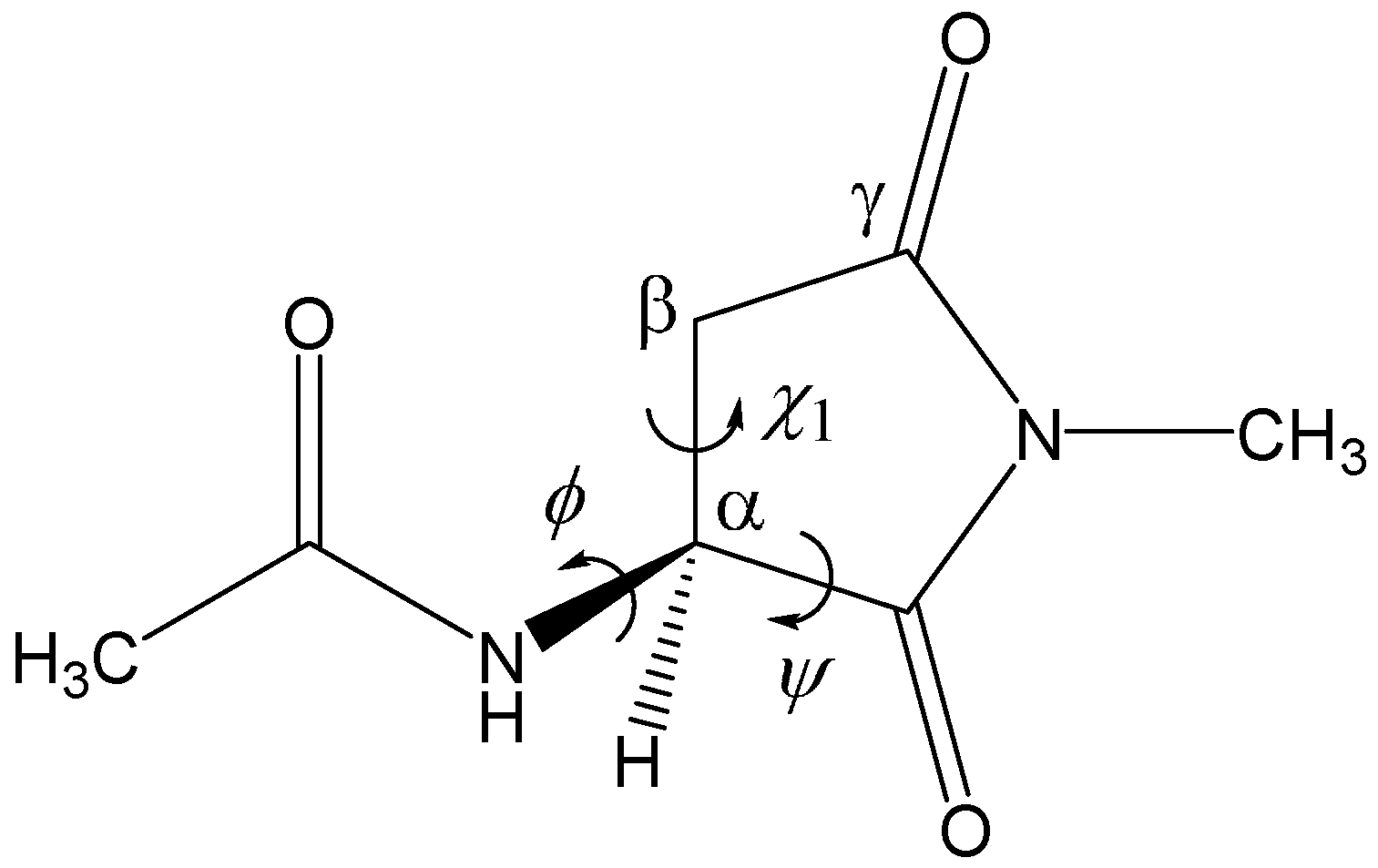
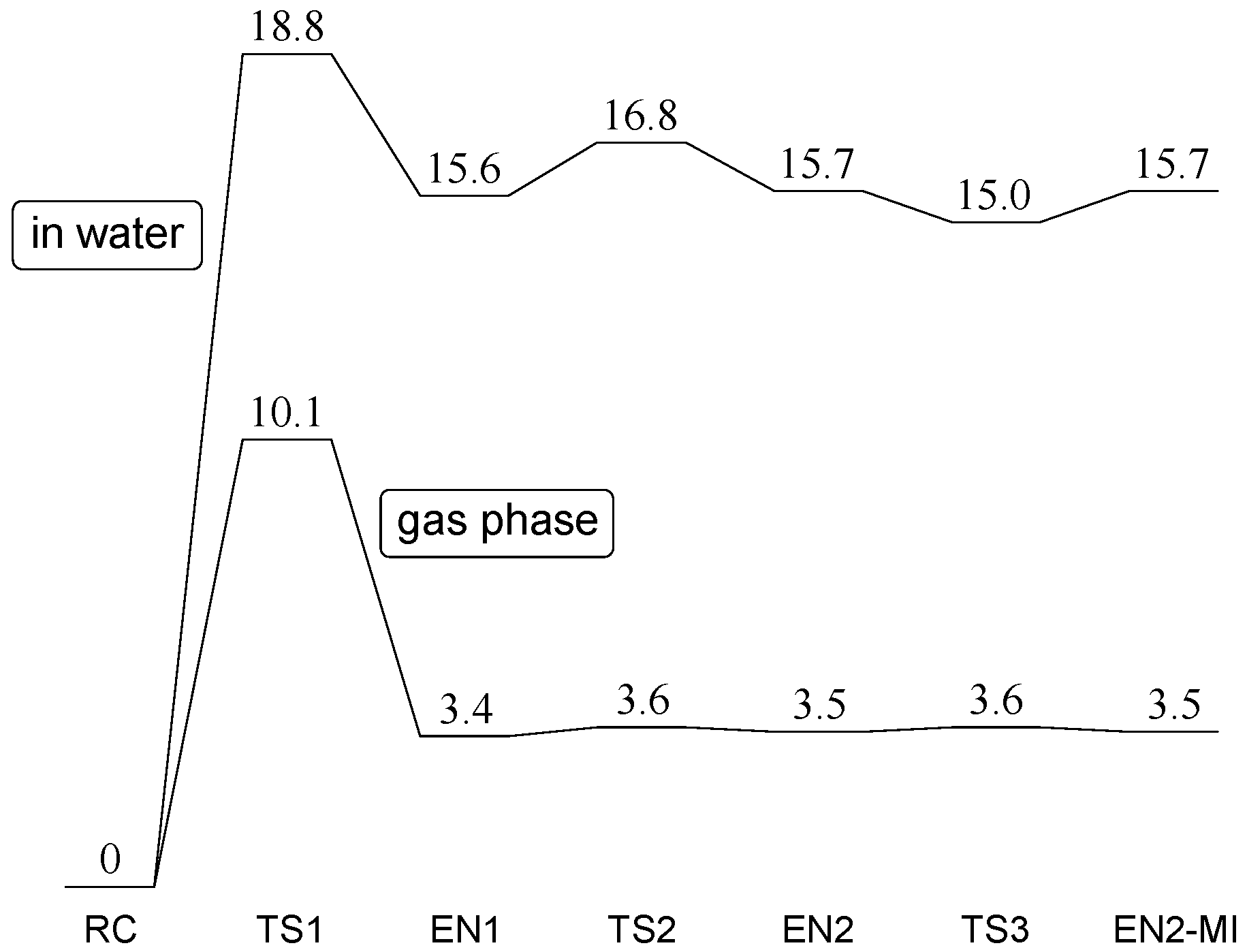
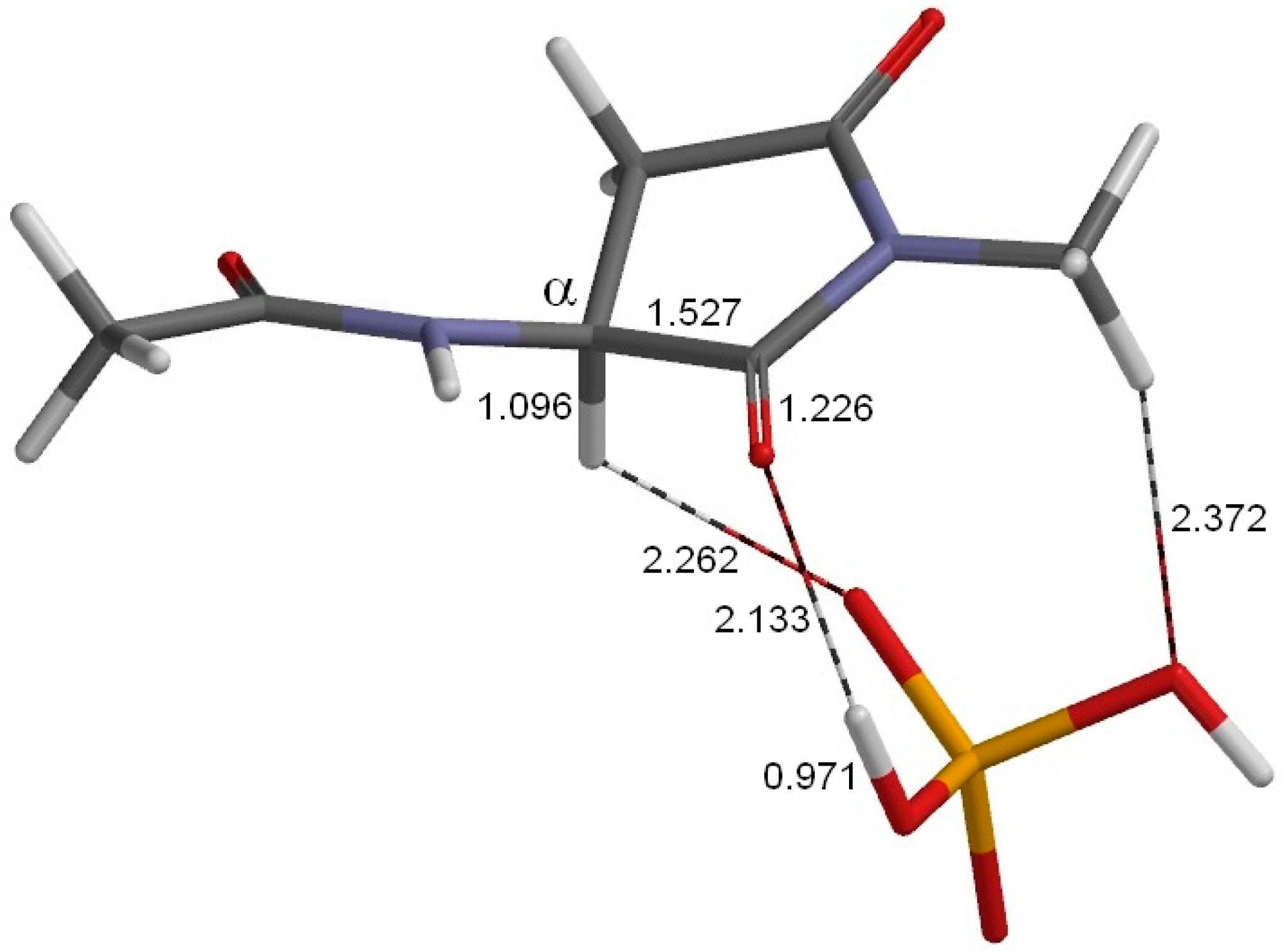
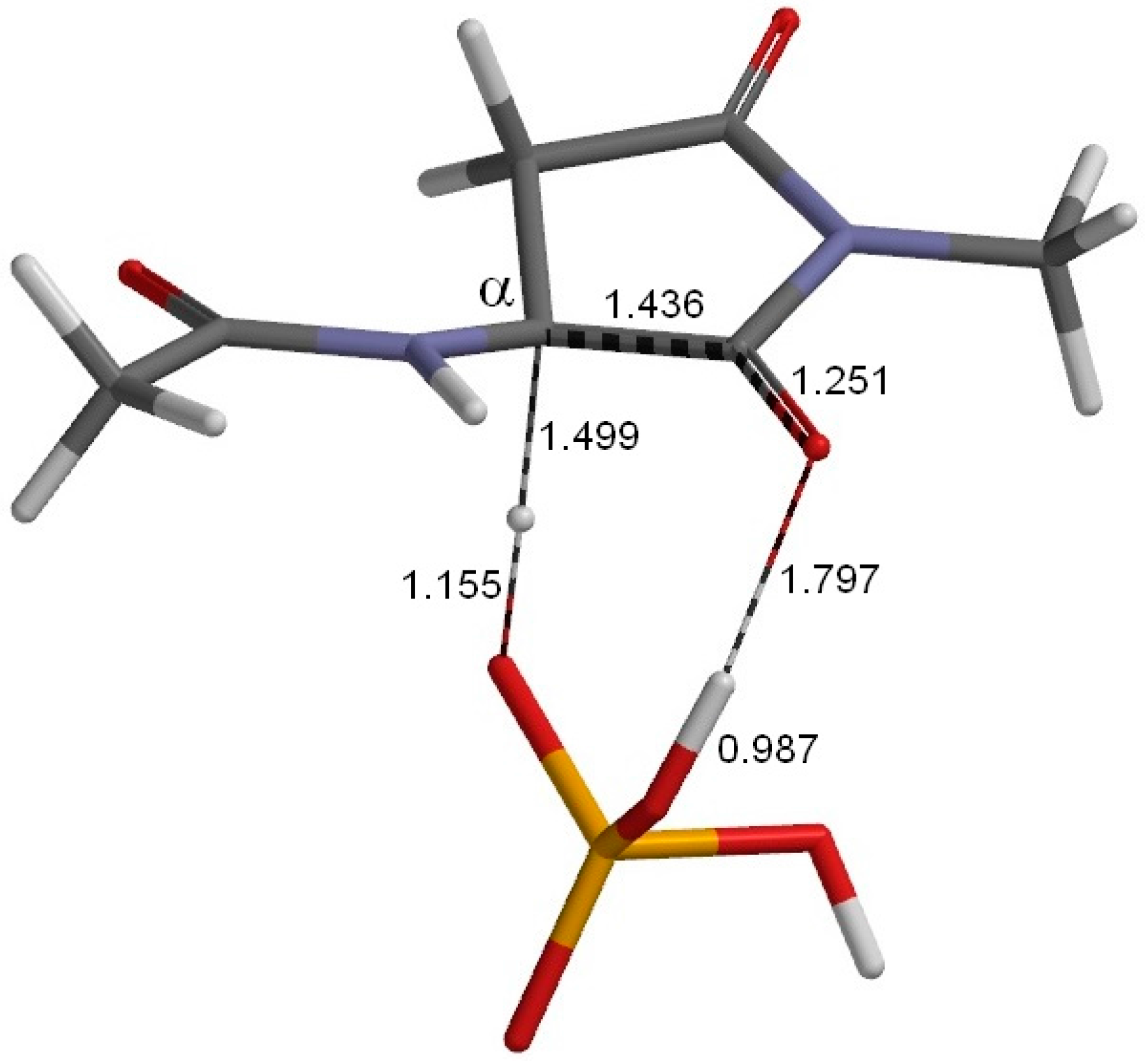
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [PubMed]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [PubMed]
- Radkiewicz, J.L.; Zipse, H.; Clarke, S.; Houk, K.N. Accelerated racemization of aspartic acid and asparagine residues via succinimide intermediates: An ab initio theoretical exploration of mechanism. J. Am. Chem. Soc. 1996, 118, 9148–9155. [Google Scholar] [CrossRef]
- Collins, M.J.; Waite, E.R.; van Duin, A.C.T. Predicting protein decomposition: The case of aspartic-acid racemization kinetics. Philos. Trans. R. Soc. Lond. B 1999, 354, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aki, K.; Fujii, N.; Fujii, N. Kinetics of isomerization and inversion of aspartate 58 of αA-crystalline peptide mimics under physiological conditions. PLoS ONE 2013, 8, e58515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, O.; Oda, A. Amide-iminol tautomerization of the C-terminal peptide groups of aspartic acid residues. Two-water-assisted mechanism, cyclization from the iminol tautomer leading to the tetrahedral intermediate of succinimide formation, and implication to peptide group hydrogen exchange. In Tyrosine and Aspartic Acid: Properties, Sources and Health Benefits; Jones, J.E., Morano, D.M., Eds.; Nova Science Publishers: New York, NY, USA, 2012; pp. 131–147. [Google Scholar]
- Takahashi, O.; Kirikoshi, R. Intramolecular cyclization of aspartic acid residues assisted by three water molecules: A density functional theory study. Comput. Sci. Discov. 2014, 7, 015005. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Roles of intramolecular and intermolecular hydrogen bonding in a three-water-assisted mechanism of succinimide formation from aspartic acid residues. Molecules 2014, 19, 11440–11452. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Acetic acid can catalyze succinimide formation from aspartic acid residues by a concerted bond reorganization mechanism: A computational study. Int. J. Mol. Sci. 2015, 16, 1613–1626. [Google Scholar] [CrossRef] [PubMed]
- Capasso, S.; Mazzarella, L.; Sica, F.; Zagari, A. Deamidation via cyclic imide in asparaginyl peptides. Pept. Res. 1989, 2, 195–200. [Google Scholar] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm. Res. 1990, 7, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. III. Effect of primary sequence on the pathways of deamidation of asparaginyl residues in hexapeptides. Pharm. Res. 1990, 7, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Tyler-Cross, R.; Schirch, V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 1991, 266, 22549–22556. [Google Scholar] [PubMed]
- Clarke, S.; Stephenson, R.C.; Lowenson, J.D. Lability of asparagine and aspartic acid residues in proteins and peptides: Spontaneous deamidation and isomerization reactions. In Stability of Protein Pharmaceuticals, Part A: Chemical and Physical Pathways of Protein Degradation; Ahern, T.J., Manning, M.C., Eds.; Plenum Press: New York, NY, USA, 1992; pp. 1–29. [Google Scholar]
- Capasso, S. Thermodynamic parameters of the reversible isomerization of aspartic residues via a succinimide derivative. Thermochim. Acta 1996, 286, 41–50. [Google Scholar] [CrossRef]
- Shapira, R.; Austin, G.E.; Mirra, S.S. Neuritic plaque amyloid in Alzheimer’s disease is highly racemized. J. Neurochem. 1988, 50, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; Greenberg, B.D. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. β-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Ishibashi, Y.; Satoh, K.; Fujino, M.; Harada, K. Simultaneous racemization and isomerization at specific aspartic acid residues in αB-crystallin from the aged human lens. Biochim. Biophys. Acta 1994, 1204, 157–163. [Google Scholar] [CrossRef]
- Fujii, N.; Satoh, K.; Harada, K.; Ishibashi, Y. Simultaneous stereoinversion and isomerization at specific aspartic acid residues in αA-crystallin from human lens. J. Biochem. 1994, 116, 663–669. [Google Scholar] [PubMed]
- Fujii, N.; Momose, Y.; Harada, K. Kinetic study of racemization of aspartyl residues in model peptides of αA-crystallin. Int. J. Pept. Protein Res. 1996, 48, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Saido, T.C.; Mann, D.M.A.; Lee, V.M.-Y.; Trojanowski, J.Q. Full-length amyloid-β(1–42(43)) and amino-terminally modified and truncated amyloid-β42(43) deposit in diffuse plaques. Am. J. Pathol. 1996, 149, 1823–1830. [Google Scholar] [PubMed]
- Fujii, N.; Takemoto, L.J.; Momose, Y.; Matsumoto, S.; Hiroki, K.; Akaboshi, M. Formation of four isomers at the Asp-151 residue of aged human αA-crystallin by natural aging. Biochem. Biophys. Res. Commun. 1999, 265, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Lowenson, J.D.; Clarke, S.; Roher, A.E. Chemical modifications of deposited amyloid-β peptides. Methods Enzymol. 1999, 309, 89–105. [Google Scholar] [PubMed]
- Orpiszewski, J.; Schormann, N.; Kluve-Beckerman, B.; Liepnieks, J.J.; Benson, M.D. Protein aging hypothesis of Alzheimer disease. FASEB J. 2000, 14, 1255–1263. [Google Scholar] [CrossRef]
- Kaneko, I.; Morimoto, K.; Kubo, T. Drastic neuronal loss in vivo by β-amyloid racemized at Ser26 residue: Conversion of non-toxic [d-Ser26] β-amyloid 1–40 to toxic and proteinase-resistant fragments. Neurosci. 2001, 104, 1003–1011. [Google Scholar] [CrossRef]
- Fujii, N.; Matsumoto, S.; Hiroki, K.; Takemoto, L. Inversion and isomerization of Asp-58 residue in human αA-crystallin from normal aged lenses and cataractous lenses. Biochim. Biophys. Acta 2001, 1549, 179–187. [Google Scholar] [CrossRef]
- Ritz-Timme, S.; Collins, M.J. Racemization of aspartic acid in human proteins. Aging Res. Rev. 2002, 1, 43–59. [Google Scholar] [CrossRef]
- Reissner, K.J.; Aswad, D.W. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 2003, 60, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N. d-Amino acid in elderly tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.L.; Collins, M.J.; Cappellini, E. Alzheimer’s disease and amyloid β-peptide deposition in the brain: A matter of “aging”? Biochem. Soc. Trans. 2010, 38, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, Y.; Konoha, K.; Kawahara, M.; Nakagomi, K. Quantification of structural alterations of l-Asp and l-Asn residues in peptides related to neuronal diseases by reversed-phase high-performance liquid chromatography. Chem. Biodivers. 2010, 7, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemisation and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. AGE 2011, 33, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Kawaguchi, T.; Sasaki, H.; Fujii, N. Simultaneous stereoinversion and isomerization at the Asp-4 residue in βB2-crystallin from the aged human eye lenses. Biochemistry 2011, 50, 8628–8635. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Sakaue, H.; Sasaki, H.; Fujii, N. A rapid, comprehensive liquid chromatography-mass spectrometry (LC-MS)-based survey of the Asp isomers in crystallins from human cataract lenses. J. Biol. Chem. 2012, 287, 39992–40002. [Google Scholar] [CrossRef] [PubMed]
- Güttler, B.H.-O.; Cynis, H.; Seifert, F.; Ludwig, H.-H.; Porzel, A.; Schilling, S. A quantitative analysis of spontaneous isoaspartate formation from N-terminal asparaginyl and aspartyl residues. Amino Acids 2013, 44, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Tambo, K.; Yamaguchi, T.; Kobayashi, K.; Terauchi, E.; Ichi, I.; Kojo, S. Racemization of the aspartic acid residue of amyloid-β peptide by a radical reaction. Biosci. Biotechnol. Biochem. 2013, 77, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Hosaka, D.; Mochizuki, N.; Akatsu, H.; Tsutsumiuchi, K.; Hashizume, Y.; Matsukawa, N.; Yamamoto, T.; Toyo’oka, T. Simultaneous determination of post-translational racemization and isomerization of N-terminal Amyloid-β in Alzheimer’s brain tissues by covalent chiral derivatized ultraperformance liquid chromatography tandem mass spectrometry. Anal. Chem. 2014, 86, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Takata, T.; Fujii, N.; Sakaue, H.; Nirasawa, S.; Takahashi, S.; Sasaki, H.; Fujii, N. Rapid survey of four Asp isomers in disease-related proteins by LC-MS combined with commercial enzymes. Anal. Chem. 2015, 87, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Takata, T.; Fujii, N. Quantitative analysis of isomeric (l-α-, l-β-, d-α-, d-β-) aspartyl residues in proteins from elderly donors. J. Pharm. Biomed. Anal. 2015, 116, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K. Isomerization of aspartyl residues in crystallins and its influence upon cataract. Biochim. Biophys. Acta 2016, 1860, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Fujii, N. Isomerization of Asp residues plays an important role in αA-crystallin dissociation. FEBS J. 2016, 283, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Konuklar, F.A.; Aviyente, V.; Sen, T.Z.; Bahar, I. Modeling the deamidation of asparagine residues via succinimide intermediates. J. Mol. Model. 2001, 7, 147–160. [Google Scholar]
- Konuklar, F.A.; Aviyente, V.; Ruiz-López, M.F. Theoretical study on the alkaline and neutral hydrolysis of succinimide derivatives in deamidation reactions. J. Phys. Chem. A 2002, 106, 11205–11214. [Google Scholar] [CrossRef]
- Konuklar, F.A.S.; Aviyente, V. Modelling the hydrolysis of succinimide: Formation of aspartate and reversible isomerization of aspartic acid via succinimide. Org. Biomol. Chem. 2003, 1, 2290–2297. [Google Scholar] [CrossRef]
- Peters, B.; Trout, B.L. Asparagine deamidation: pH-dependent mechanism from density functional theory. Biochemistry 2006, 45, 5384–5392. [Google Scholar] [CrossRef] [PubMed]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Reaction mechanism of deamidation of asparaginyl residues in peptides: Effect of solvent molecules. J. Phys. Chem. A 2006, 110, 8354–8365. [Google Scholar] [CrossRef] [PubMed]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 2009, 113, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Kobayashi, K.; Oda, A. Modeling the enolization of succinimide derivatives, a key step of racemization of aspartic acid residues: Importance of a two-H2O mechanism. Chem. Biodiv. 2010, 7, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, H.; Yamada, H.; Wada, K.; Imoto, T. Stabilization of lysozyme against irreversible inactivation by suppression of chemical reactions. J. Biochem. 1995, 117, 635–640. [Google Scholar] [PubMed]
- Bohne, C.; MacDonald, I.D.; Dunford, H.B. Measurement of rates and equilibria for keto-enol tautomerism of aldehydes using horseradish peroxidase compound I. J. Am. Chem. Soc. 1986, 108, 7867–7868. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.P.; Bez, G. Henry reaction in aqueous media at neutral pH. Eur. J. Org. Chem. 2013, 2922–2929. [Google Scholar] [CrossRef]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Wavefunction Inc. Spartan ’14, version 1.1.4; Wavefunction, Inc.: Irvine, CA, USA, 2014.
- Takahashi, O. Two-water-assisted racemization of the succinimide intermediate formed in proteins. A computational model study. Health 2013, 5, 2018–2021. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of the Succinimide Intermediate Formed in Proteins and Peptides: A Computational Study of the Mechanism Catalyzed by Dihydrogen Phosphate Ion. Int. J. Mol. Sci. 2016, 17, 1698. https://doi.org/10.3390/ijms17101698
Takahashi O, Kirikoshi R, Manabe N. Racemization of the Succinimide Intermediate Formed in Proteins and Peptides: A Computational Study of the Mechanism Catalyzed by Dihydrogen Phosphate Ion. International Journal of Molecular Sciences. 2016; 17(10):1698. https://doi.org/10.3390/ijms17101698
Chicago/Turabian StyleTakahashi, Ohgi, Ryota Kirikoshi, and Noriyoshi Manabe. 2016. "Racemization of the Succinimide Intermediate Formed in Proteins and Peptides: A Computational Study of the Mechanism Catalyzed by Dihydrogen Phosphate Ion" International Journal of Molecular Sciences 17, no. 10: 1698. https://doi.org/10.3390/ijms17101698
APA StyleTakahashi, O., Kirikoshi, R., & Manabe, N. (2016). Racemization of the Succinimide Intermediate Formed in Proteins and Peptides: A Computational Study of the Mechanism Catalyzed by Dihydrogen Phosphate Ion. International Journal of Molecular Sciences, 17(10), 1698. https://doi.org/10.3390/ijms17101698