1. Introduction
Hepatitis C virus (HCV) is a positive-stranded RNA virus ~9.6 kb in length, and it belongs to the genus
Hepacivirus within the
Flaviviridae family [
1]. It is estimated that 170 million people worldwide are chronically infected with HCV and at risk of liver fibrosis, cirrhosis and hepatocellular carcinoma [
2]. The HCV genome encodes at least three structural proteins: core and two envelope glycoproteins (E1 and E2); and seven nonstructural (NS) proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B).
Globally, at least seven HCV genotypes and 67 subtypes have been characterized. HCV isolates differ by >15% among different genotypes or subtypes [
3]. HCV genotypes and subtypes are distributed differently among different areas of the world. In Japan, HCV genotype 1b is the major genotype (70%), followed by HCV genotype 2a (20%) and 2b (10%) [
4,
5]. Several methods have been established to determine HCV genotypes, such as sequence analysis [
6], restriction fragment length polymorphism [
7], hybridization of PCR products with specific probes [
8] and next-generation sequencing [
5].
Accurate determination of the HCV genotypes is essential for the selection of proper antiviral drugs and treatment regimens in HCV-infected individuals [
9]. Treatment response also depends on HCV subgenotypes [
10]. In Japan, the choice of antiviral regimen against HCV is generally based on the results of HCV serotyping as the national health insurance system currently approves only HCV serotyping methods [
11]. Although previous reports indicate that the detection rate of serotyping is generally high and the risk of misdiagnosis is considered rare [
11], we have experienced few cases with misidentified genotypes in the treatment of hepatitis C. It is important to elucidate the mechanism responsible for the discrepancy between HCV genotyping based on directly sequencing of the HCV genome and HCV serotyping by enzyme immunoassay (EIA).
In the present study, we described cases with discrepant results between HCV genotyping and serotyping assays that were based on the 5′-untranslated region (5′-UTR) and NS4 regions, respectively. Sanger sequencing was performed for the HCV core and NS4 regions, and deep sequencing was conducted for HCV NS4 regions to study the validity of the serotyping and genotyping results.
3. Discussion
The distribution of HCV genotypes is different among individuals with different infection routes such as blood transfusion, intravenous drug use and tattooing. In Japan, HCV genotype 1b had been associated with blood transfusion, whereas genotypes 2a and 2b were associated with injections or tattooing [
15]. In the present study, 14 of 18 patients were confirmed as HCV genotype 2, supporting the previous report that described that the NS4-based serotyping assay had lower concordance for genotype 2b specimens [
16]. Among these 14 patients, seven (50%) had a history of intravenous injection or tattooing. These patients are considered to be multiple HCV-exposed individuals, and some of them might have been exposed to mixed infection with different HCV genotypes and subtypes.
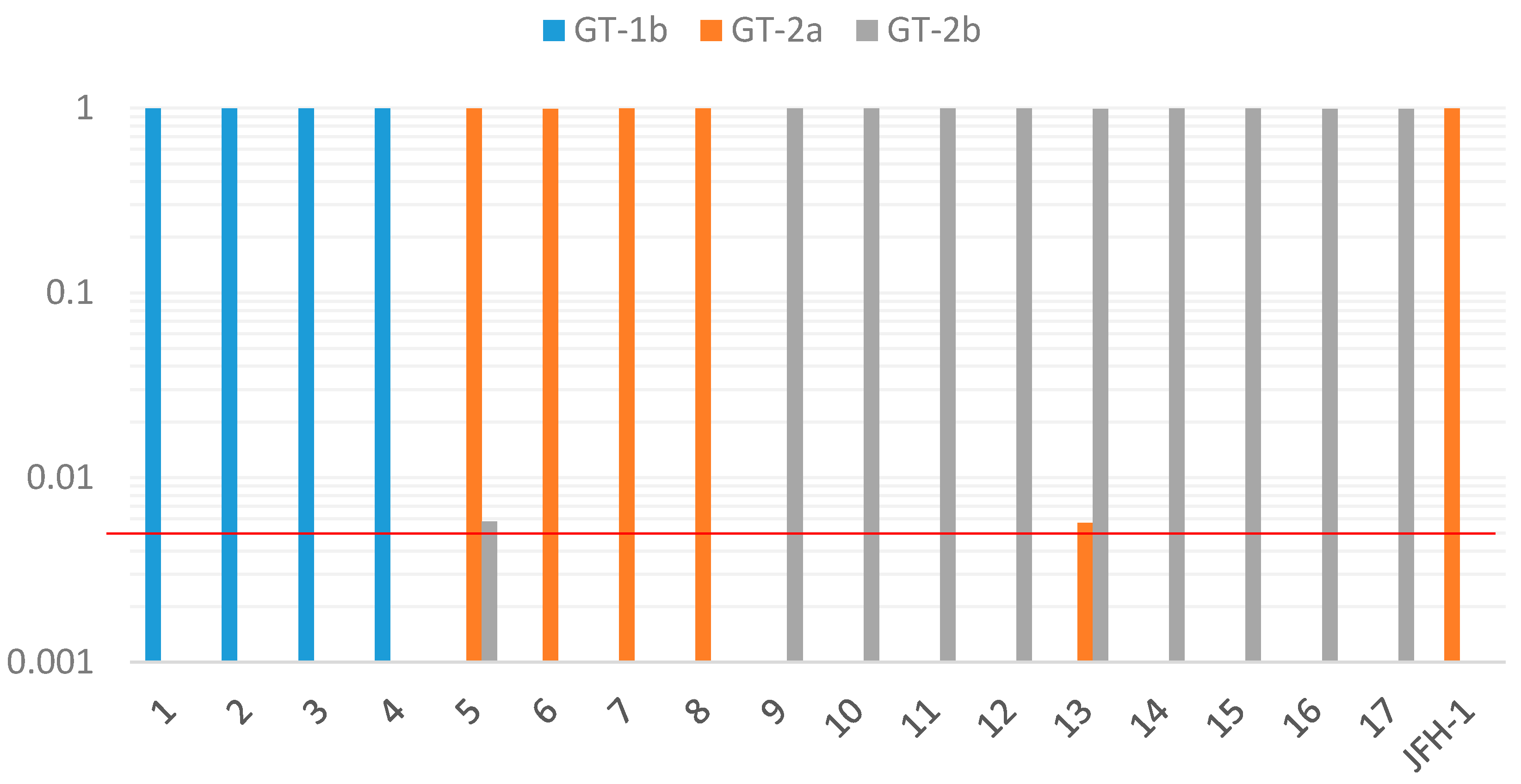
In the present study, the same genotype was detected by cloned Sanger sequencing at the core and NS4 regions of the HCV genome as the genotype determined by 5′-UTR. No case was detected as mixed infection. As the possibility of the coexistence of a few minor variants could not be completely ruled out because only five clones were analyzed in this study, we further examined this possibility by the deep sequencing method. Deep sequencing of the HCV NS4 region produced large sequence depth ranging from 45 to 993 thousands of reads from each sample. In all 17 cases of the present study, more than 99% of the reads were assigned to the same genotype as determined by other genotyping methods. Furthermore, in two of the 17 samples, a minor population of sequences with different HCV subgenotypes was detected, suggesting the possibility of mixed infection in these samples. It is difficult to detect minor variants, which is between 0.5% and 1%, by the standard cloning method. Deep sequencing could rapidly and easily detect minor variants with a large sequence depth compared to the cloning method. As for sequencing error, our study using the JFH1 clone revealed that up to 0.4% of reads were assigned to incorrect genotype (2b), consistent with a previous report describing a sequencing error rate of below 0.4% in the Illumina platform [
17]. However, Thomson et al. [
18] suggested that the detection of minority variants is less reliable at lower ratios. The cases of mixed infection with different HCV genotypes, as well as the recombinant forms of HCV are very rare, even in a highly exposed group, such as intravenous drug users (IVDU) [
19].
Among the 18 cases, 11 were confirmed to show different HCV serotyping results from HCV genotyping; two cases showed HCV genotype 1b with HCV serotype 2; while nine cases showed HCV genotype 2 with HCV serotype 1. Compared to the core region, nucleotide sequence variations in the NS4 region were more commonly existing between samples within the same genotype (
Table 3 and
Table 4). We examined the possibility that variations of the amino acid sequence could affect the serotyping results. Both consensus sequence (
Table 5A) and cloning analysis in the NS4 region (
Table 5B,C) showed that although several amino acid variations existed when compared with the HCV genotype-specific reference peptide, each sequence was much closer to the reference peptide with the same genotype. It should be noted that the error rate of Taq DNA polymerase is approximately 2 × 10
−5, estimating that one nucleotide error could appear in 50% of the amplified DNA molecules by nested PCR. Furthermore, comparative analysis with the control group will be needed to better understand the mechanism of discrepancy.
Among the 18 HCV specimens serotyped in our series, four samples (22.2%) were defined as “not determined”. It has been suggested that HCV serotyping analysis provides an indirect typing method based on the production of type-specific antibodies by the infected host. Therefore, the sensitivity of HCV serotyping depends on the immune response of the HCV-infected host. Therefore, the possibility that some of the patients with HCV infection did not produce antibody against NS4 protein should be considered. By the HCV serotyping assay, two of the 18 patients (11.1%) had mixed reactions. Multiple exposures to different HCV strains could be one possible mechanism for these findings, although the possibility of non-specific reaction also exists. For such cases, instead of HCV serotyping, HCV genotype testing can be a good alternative method.
A major limitation of our study is that we could not describe the sensitivity and concordance between HCV serotyping and genotyping data in the entire cohort due to lack of a comparable group. Previous studies [
11,
20] using the same NS4 antigen (C14) as ours showed that the sensitivity of serotypes 1 and 2 is 95.8%–100%, and there was no discordant case. We repeated the initial serotyping assay using the same antigen with the newly-updated chemiluminescent EIA system (HISCL HCV Gr reagents, Sysmex, Japan). According to the manufacturer’s instruction, the sensitivity of the new system is 84.4%, and the serotyping results showed 100% concordance with those of the older system. Our re-serotyping analysis showed that new system could correctly detect type 2-specific antibody in six samples (Nos. 6, 8, 10, 12, 17 and 18) and type 1 antibody in two samples (Nos. 2 and 4). However, it could not detect any antibodies in eight cases, showed the incorrect serotype in one case (No. 14) and mixed serotype in one case (No. 11). These results suggest that comparing different serotyping systems would be useful to understand the discrepant cases further.
After the identification of serotype-specific linear epitopes in the core and NS4 region [
11,
14], several serotyping systems have been reported using peptides corresponding to these regions. Besides the C14 serotyping system we used, the MUREX HCV Serotyping 1–6 assay (Abbott Diagnostics) has been developed [
21] using type-specific NS4 antigen corresponding to HCV types 1–6. Prescott et al. [
22] reported that 15 discrepant cases (151/166) could be detected using the old version of this assay (HC02). According to the instructions, the concordance (96.12%) of the new version (2G26) becomes higher than an earlier version (HC02) (90.37%), while the sensitivity of serotypes 1 and 2 is 76.8% and 75.5% in the new version. The RIBA HCV serotyping assay (Chiron Corporation) [
16,
23,
24] detected anti-HCV antibodies against five synthetic peptides from the NS4 region and three peptides from the core region, and it has been reported that the sensitivity/concordance of serotypes 1 and 2 is 84.7%–96.5%/96.2%–100% and 82.4%–100%/93.4%–100%, respectively. Taken together, serotyping generally shows high concordance with genotyping. However, attention should be paid to the fact that there could occur cases with no response to the antigen and also with discrepancy even if they can be regarded as uncommon events based on previous studies.
Only sequence analysis of specific gene regions of the HCV genome that are predictive of HCV genotype was completely reliable for HCV genotyping [
25]. There are several genotyping methodologies available, such as sequencing, primer-specific PCR, real-time PCR and the line probe assay. Chantratita et al. [
26] compared two PCR-based line probe assays and recently reported that the six HCV genotyping 9G test showed overall sensitivity and specificity higher than 92.5% and 99.4%, respectively. On the other hand, this method based on PCR may not be correctly estimated in cases with low-level viremia, such as No. 18 in this study, or if serum samples have not been stored correctly [
27]. Meanwhile, one sample (No. 17) could not be genotyped by the HCV genotype-specific PCR method. In this case, even small sequence variations in the primer region could affect the results due to high sequence similarity in this region (
Table 3).
Serological methods are based on the detection of antibodies against HCV serotype-specific epitopes and are easier and faster for determining HCV serotypes. It is possible that HCV serotyping may allow the determination of HCV types of both past and present infection. Moreover, its high performance on samples with low viral load offers an obvious advantage over the genotyping methods. The cost of the HCV serotyping method is less than that of HCV genotyping. ELISA of the HCV serotyping method is seldom associated with the risk of contamination. Thus, HCV serotyping is particularly suitable for epidemiologic studies.
Treatment outcomes of patients are shown in
Table 7. In two of four patients with HCV genotype 1b, treatment with daclatasvir plus asunaprevir for 24 weeks could lead to sustained virological response at 24 weeks after the stoppage of treatment (SVR24). In four of five patients with HCV genotype 2a, treatment with sofosbuvir plus ribavirin was performed for 12 weeks, and all four patients achieved SVR24. Among the total of nine patients with HCV genotype 2b, five were treated with interferon-free or interferon-including regimens and achieved SVR24. Although six patients (Nos. 1, 2, 3, 4, 9, 16) had been treated with interferon-including treatments without DAAs based on serotyping results, some patients (Nos. 2, 4, 9), developed relapse after completing the treatment course, and others (Nos. 1, 3, 16) had no response.
Better treatment outcome can be obtained when the treatment is based on HCV genotyping results. For example, Patient No. 2 had been a candidate for sofosbuvir and ribavirin regimen based on the serotyping results (serotype 2), but the patient was treated with daclatasvir and asunaprevir regimen after getting the genotyping result of genotype 1b and achieved SVR24. Sohda et al. [
28] reported the non-response to daclatasvir and asunaprevir therapy in patients coinfected with hepatitis C virus genotypes 1 and 2.



 ,
, 

{kind=link}