
The Regulatory Capacity of Bivalent Genes—A Theoretical Approach


Abstract
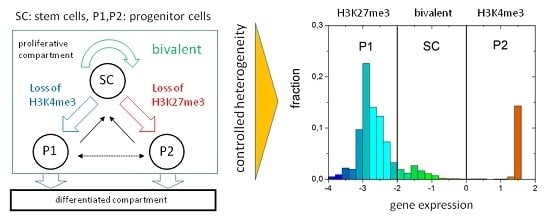
:
1. Introduction
2. Basic Model Assumptions
2.1. Epigenetic Regulation of Transcription: The Basic Regulatory Circuit
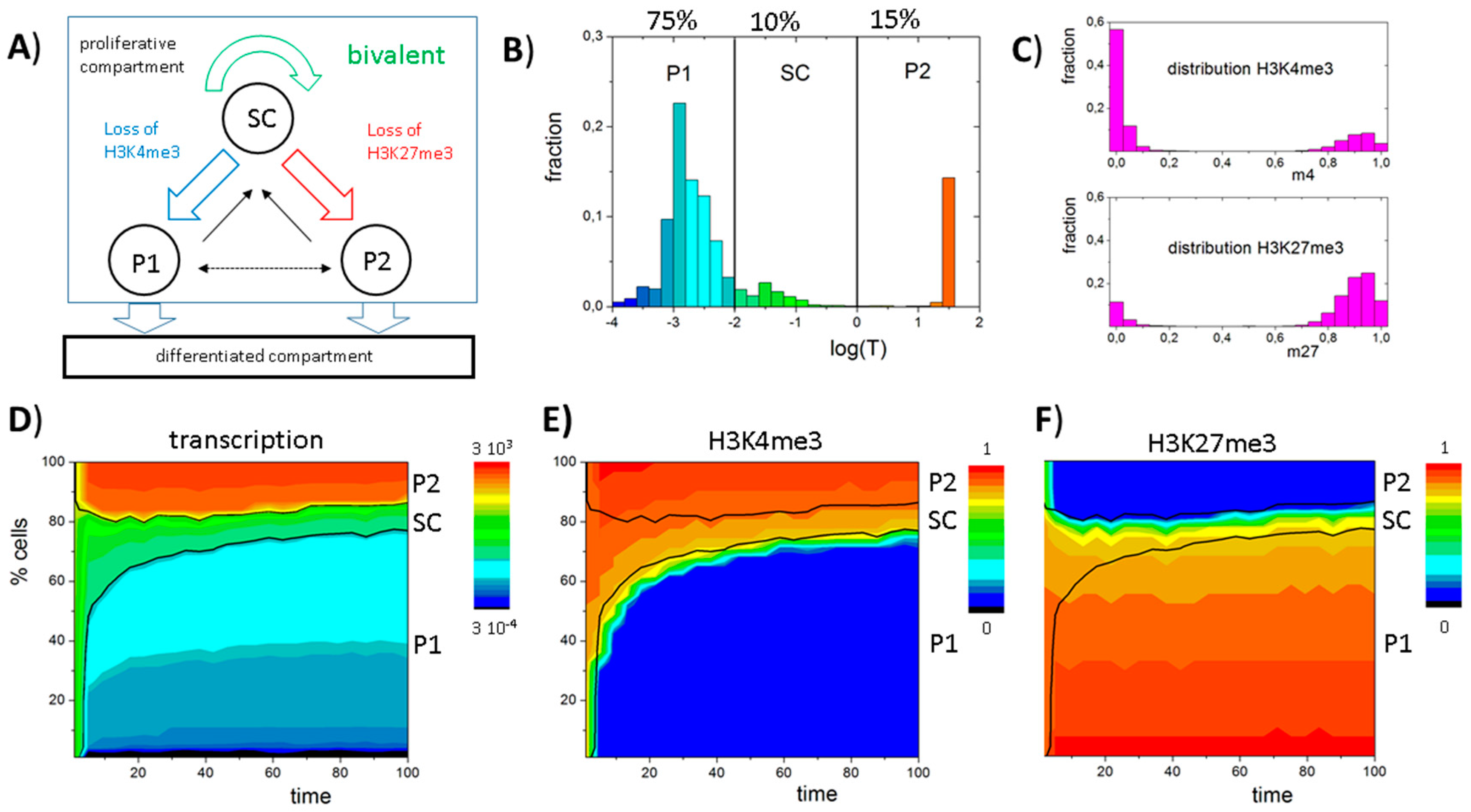
2.2. Simulations of Cell Populations
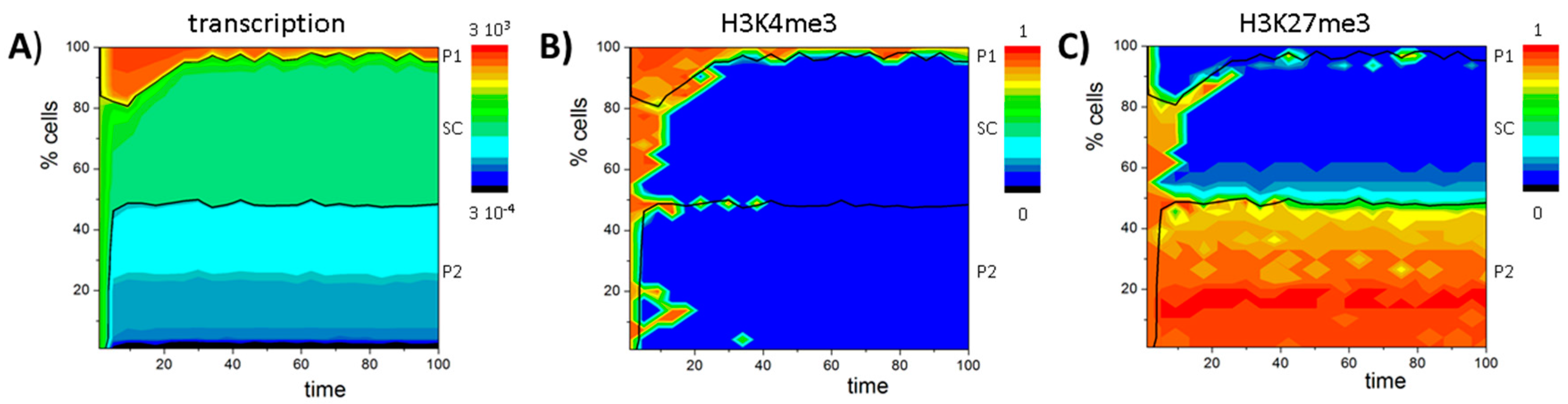
3. Results
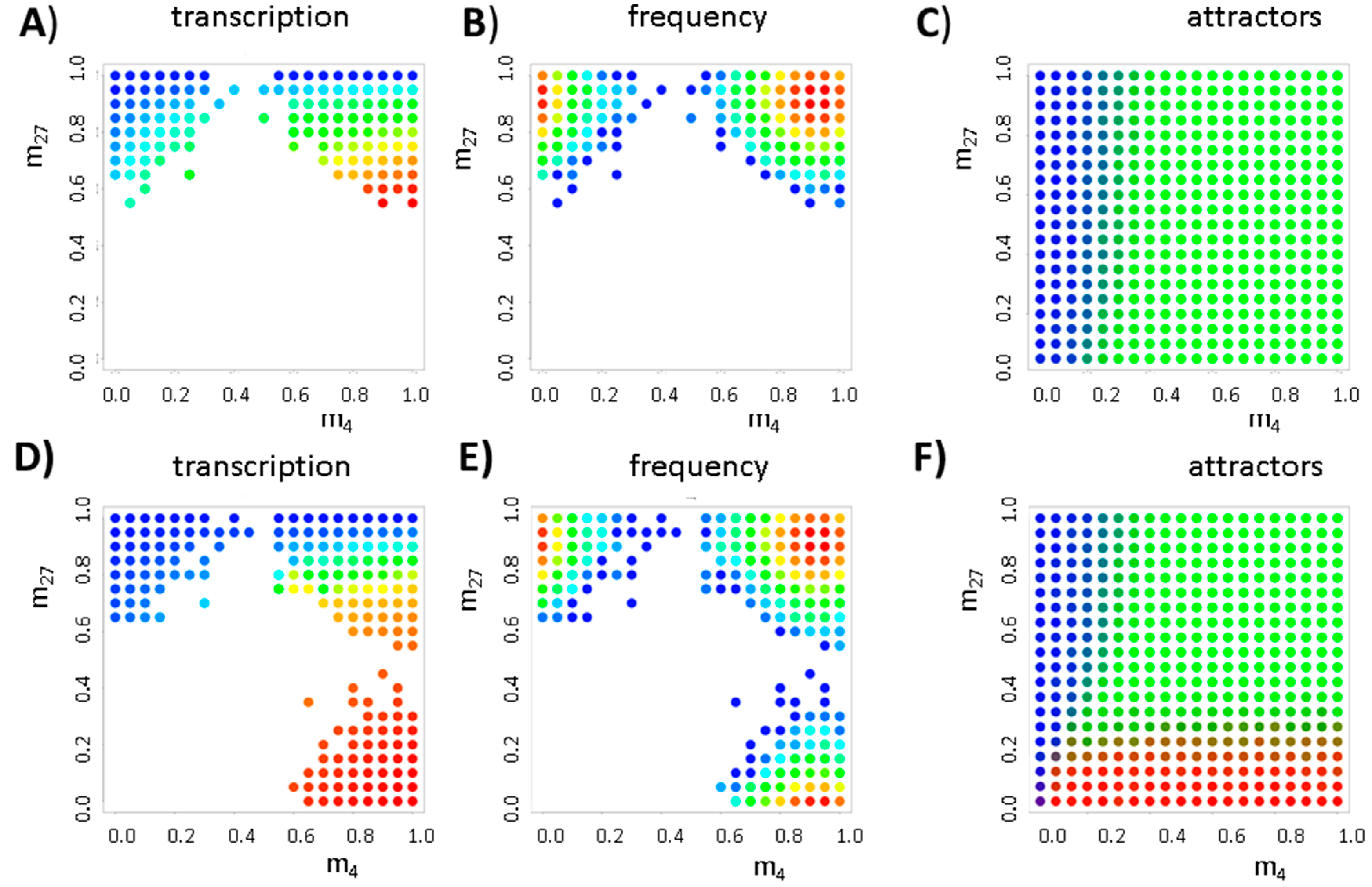
3.1. The Occurrence of the Bivalent Modification States
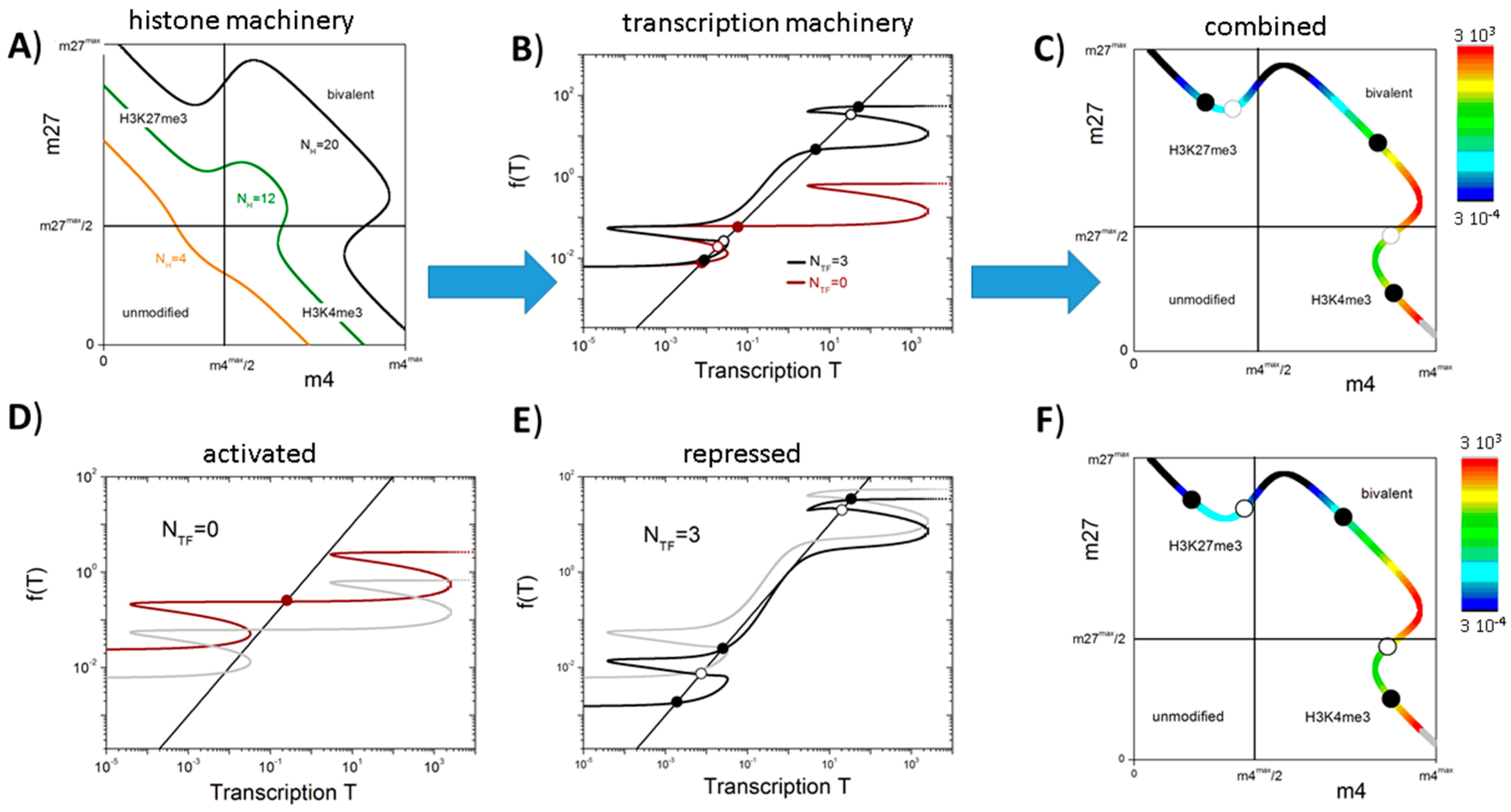
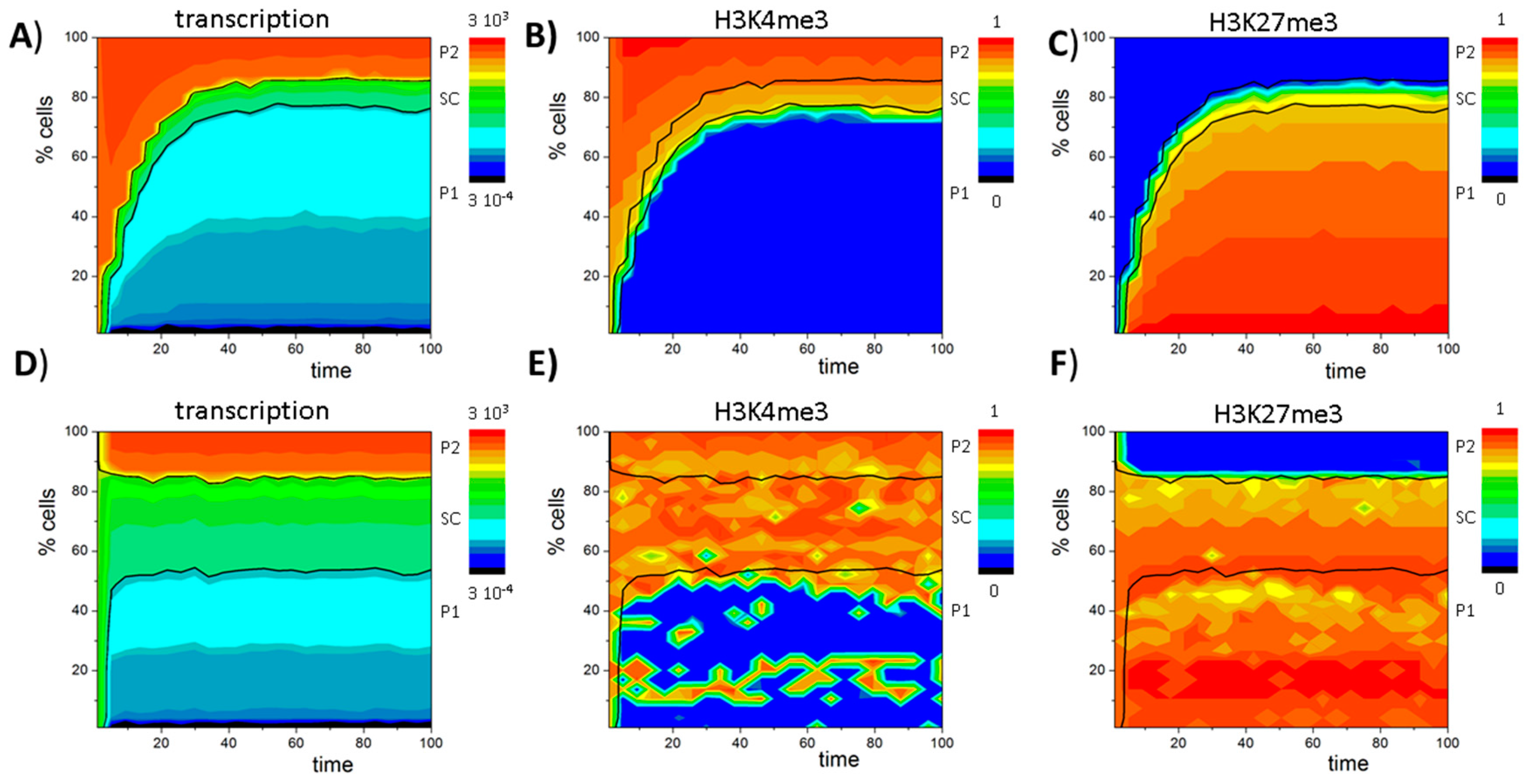
3.2. Balance between Histone Modification States (the Histone Modification Machinery)
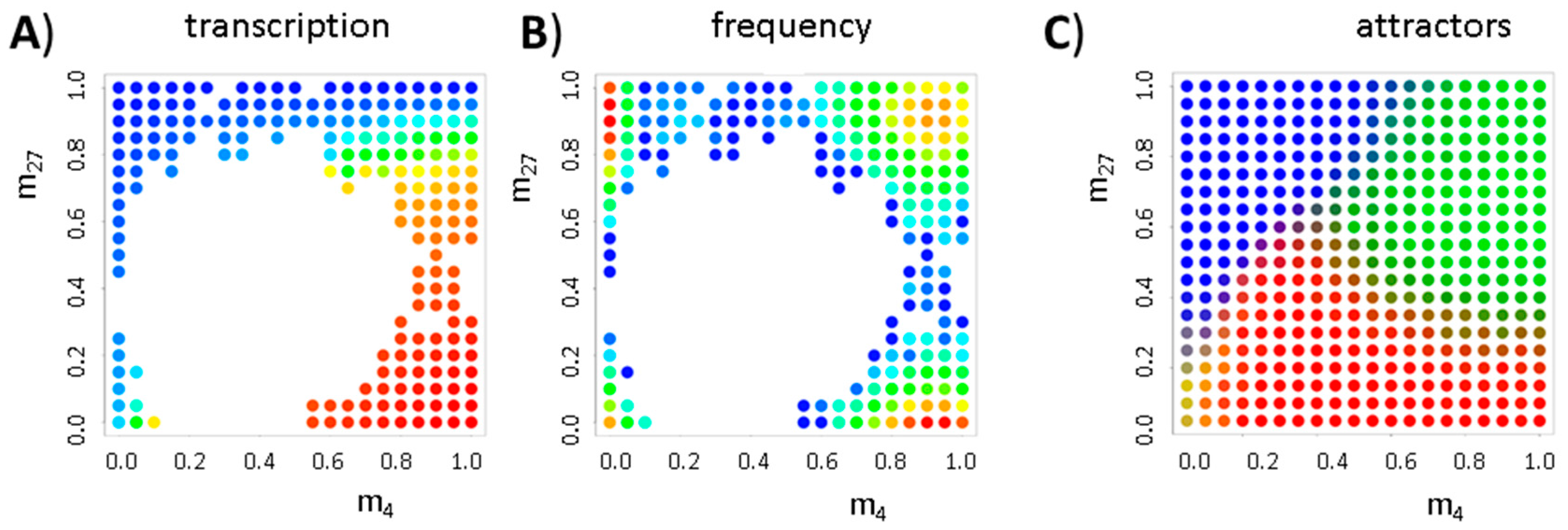
3.3. Transcription Controls Epigenetics (the Transcriptional Machinery)
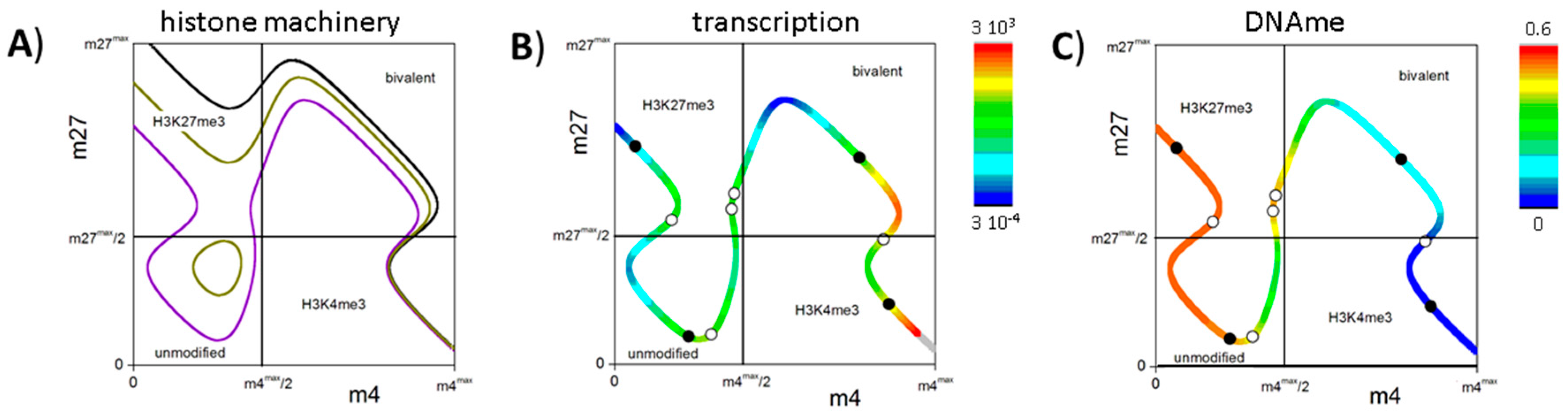
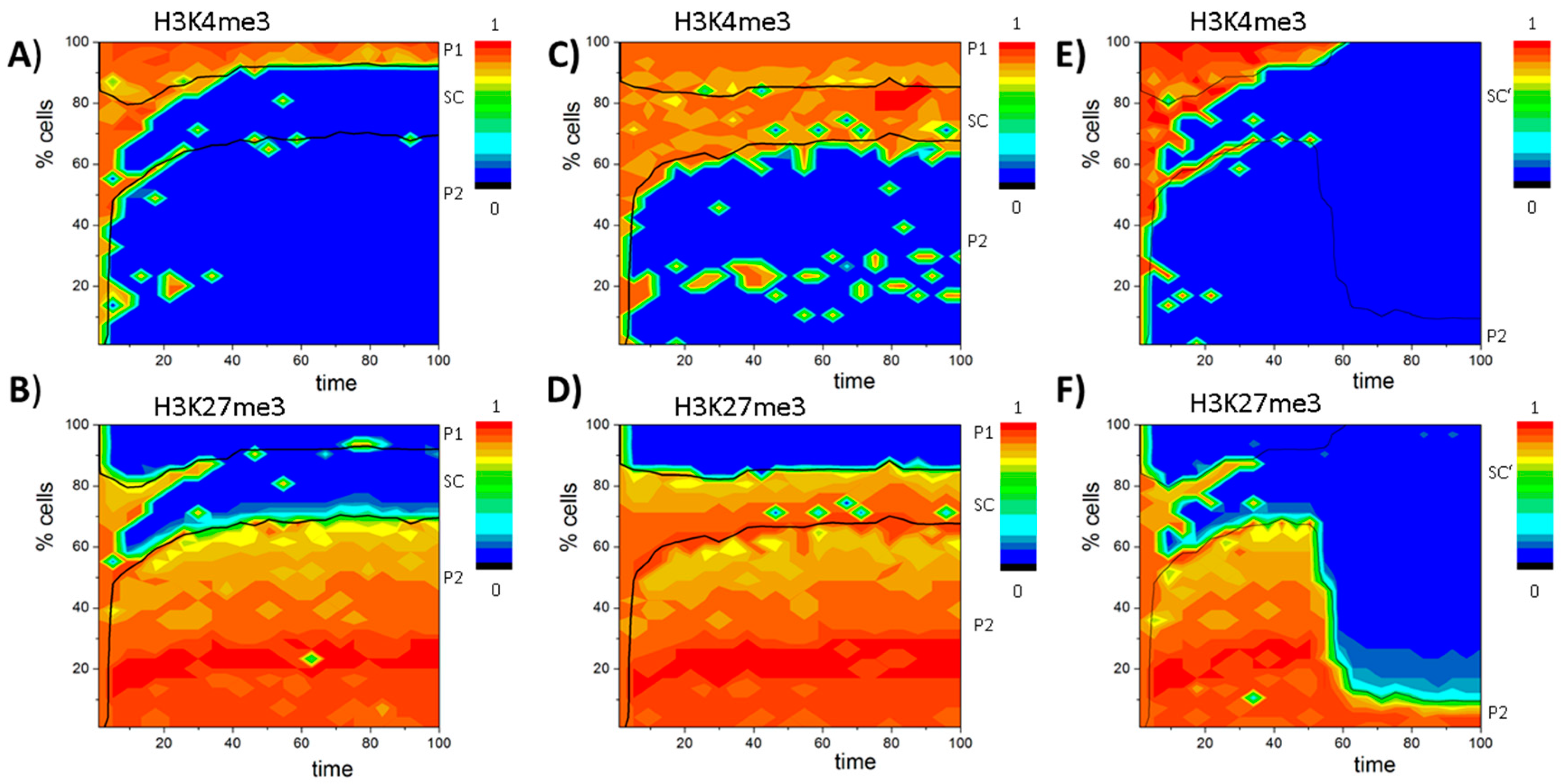
3.4. Histone Modification Can Instruct Gene Expression
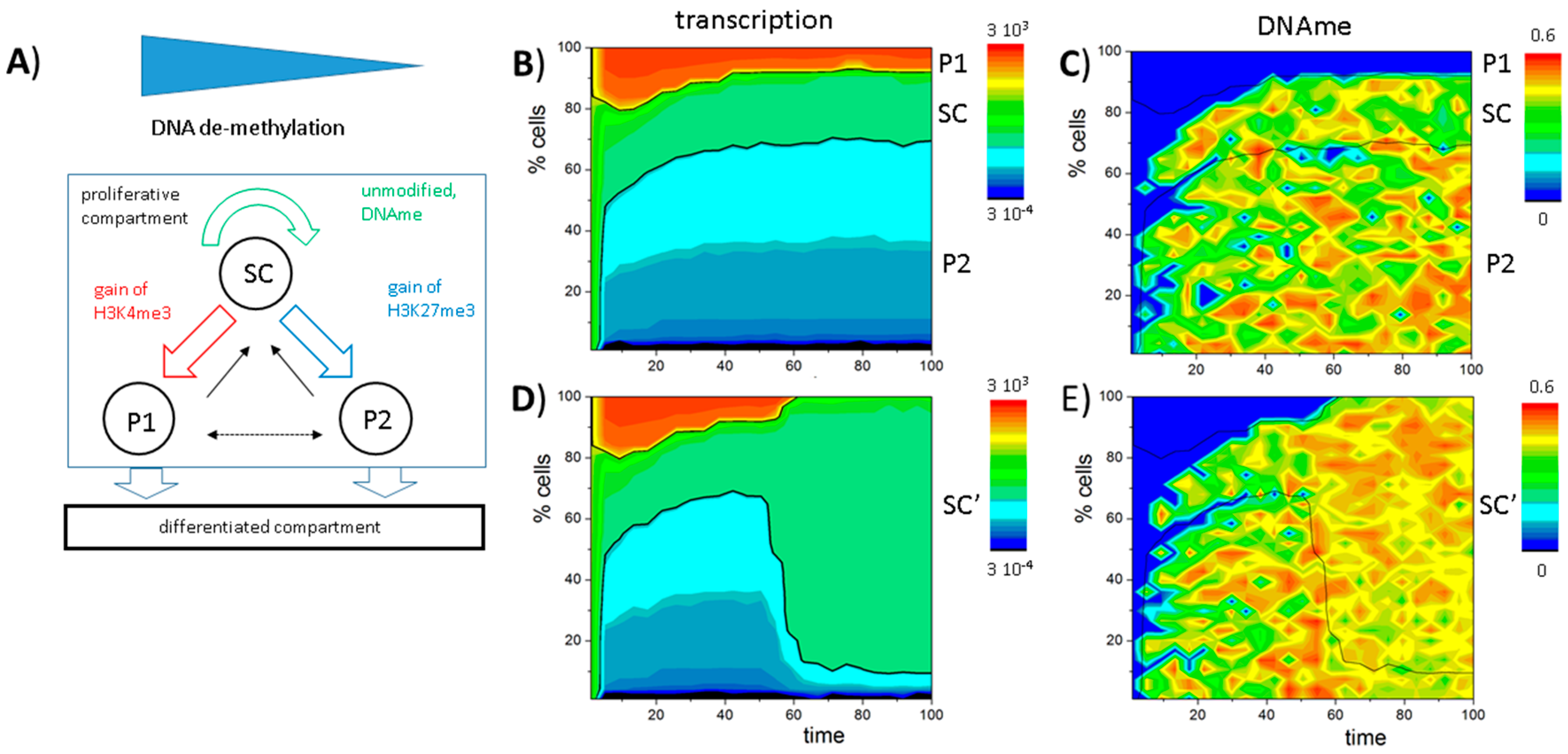
3.5. DNA Methylation Destabilizes Bivalent States
3.6. A Model of Blast Formation during AML
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| HMT | histone methyltransferase |
| DNMT | DNA methyltransferase |
| TF | transcription factor |
| MV1 | model version 1 |
| MV2 | model version 2 |
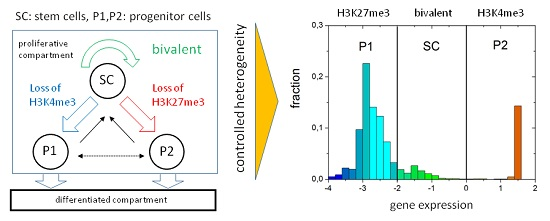
Appendix A. Model of Histone Modification
Regulation of Histone Modification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Description |
|---|---|---|
| ε04, ε027 | 9.0, 10.0 | ground enthalpy per bound HMT * |
| εBS4, εBS27 | −5.0, −5.0 | free energy of CpG binding * |
| εHM4, εHM27 | −0.5, −0.5 | free energy of histone binding * |
| C4, C27 | 0.1, 0.1 | de-modification constant |
| NH | 4/12/20 | number of cooperative nucleosomes |
Appendix B. Model of Transcriptional Regulation
Regulation of Gene Expression
| Parameter | Value | Description |
|---|---|---|
| Pmax | 100 | maximum transcription rate |
| δ | 1.5 | transcript degradation rate |
| εA | 2 | free energy of polymerase binding * |
| FTF | 4/1/0.25 | regulation factor of the TF-network |
| NTF | 0/3 | number of binding sites for auto-activation |
Appendix C. Stochastic Simulation


Appendix D. The GATA3 Circuit


Appendix E. DNA Methylation Model
| Parameter | Value | Description |
|---|---|---|
| Dmain | 0.8 | probability of maintaining DNA methylation |
| Dnovo | 0.0/0.1/0.3 | probability of de novo DNA methylation |
| DCG4, DCG27 | 6, 4 | interaction energy between HMTs and DNMTs * |
| DDE0 | 6/2 | maximum rate of active DNA demethylation |
Appendix F. The PU.1 Circuit


References
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zang, C.; Roh, T.Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Steiner, L.; Przybilla, J.; Rohlf, T.; Prohaska, S.; Galle, J. Transcriptional regulation by histone modifications: Towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 2013, 10, 26006. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; LeRoy, G.; Drury, W.J., III; Zee, B.M.; Son, J.; Beck, D.B.; Young, N.L.; Garcia, B.A.; Reinberg, D. Asymmetrically modified nucleosomes. Cell 2012, 151, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Block, K.F.; Pasini, A.; Baylin, S.B.; Easwaran, H. Genome-wide positioning of bivalent mononucleosomes. BMC Med. Genom. 2016, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.J.; Cui, K.; Tang, Q.; Zhao, K. Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genom. 2013, 14, 193. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Kazmierczak, C.; Duchêne, B.; Jonckheere, N.; Leteurtre, E.; van Seuningen, I. Cryosectioning the intestinal crypt–villus axis: An ex vivo method to study the dynamics of epigenetic modifications from stem cells to differentiated cells. Stem Cell Res. 2015, 14, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Stojanova, Z.P.; Kwan, T.; Segil, N. Epigenetic regulation of ATOH1 guides hair cell development in the mammalian cochlea. Development 2016, 143, 1632. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Rohlf, T.; Loeffler, M.; Galle, J. Understanding epigenetic changes in aging stem cells—A computational model approach. Aging Cell 2014, 13, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Buske, P.; Binder, H.; Galle, J. Histone modifications control DNA methylation profiles during ageing and tumour expansion. Front. Life Sci. 2013, 7, 31–43. [Google Scholar]
- Buratowski, S.; Kim, T. The role of cotranscriptional histone methylations. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 95–102. [Google Scholar] [PubMed]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.; van Schaik, F.M.; Varier, R.A.; Baltissen, M.P.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Jermann, P.; Hoerner, L.; Burger, L.; Schübeler, D. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3415–E3421. [Google Scholar] [PubMed]
- Pasini, D.; Malatesta, M.; Jung, H.R.; Walfridsson, J.; Willer, A.; Olsson, L.; Skotte, J.; Wutz, A.; Porse, B.; Jensen, O.N.; et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of polycomb group target genes. Nucleic Acids Res. 2010, 38, 4958–4969. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.; Deaton, A.; Andrews, R.; James, K.D.; et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010, 464, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.; Appanah, R.; Lee, S.; Lam, L.L.; Goyal, P.; Lorincz, M.C. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of DNMT3a but not de novo DNA methylation. Epigenetics 2009, 4, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Mutskov, V.; Felsenfeld, G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 2004, 23, 138–149. [Google Scholar] [PubMed]
- Dar, R.D.; Razooky, B.S.; Singh, A.; Trimeloni, T.V.; McCollum, J.M.; Cox, C.D.; Simpson, M.L.; Weinberger, L.S. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl. Acad. Sci. USA 2012, 109, 17454–17459. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Barth, T.K.; Reverón-Gómez, N.; Sidoli, S.; Schmidt, A.; Jensen, O.N.; Imhof, A.; Groth, A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015, 29, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Galle, J.; Loeffler, M.; Drasdo, D. Modeling the effect of deregulated proliferation and apoptosis on the growth dynamics of epithelial cell populations in vitro. Biophys. J. 2005, 88, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.; Buetow, K.H.; et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Huang, G.; Rosenbauer, F.; Evans, E.K.; Radomska, H.S.; Iwasaki, H.; Akashi, K.; Moreau-Gachelin, F.; Li, Y.; Zhang, P.; et al. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol. Cell. Biol. 2005, 25, 2832–2845. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Junker, J.P.; Mokry, M.; Hatzis, P.; Koo, B.K.; Sasselli, V.; van der Flier, L.G.; Cuppen, E.; van Oudenaarden, A.; Clevers, H. ASCL2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015, 16, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, H.; Sigvardsson, M.; Attema, J.L. Epigenetic chromatin states uniquely define the developmental plasticity of murine hematopoietic stem cells. Blood 2010, 115, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Attema, J.L.; Papathanasiou, P.; Forsberg, E.C.; Xu, J.; Smale, S.T.; Weissman, I.L. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12371–12376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ouyang, W. The function role of GATA-3 in Th1 and Th2 differentiation. Immunol. Res. 2003, 28, 25–37. [Google Scholar] [CrossRef]
- Van de Walle, I.; Dolens, A.C.; Durinck, K.; De Mulder, K.; Van Loocke, W.; Damle, S.; Waegemans, E.; de Medts, J.; Velghe, I.; de Smedt, M.; et al. GATA3 induces human T-cell commitment by restraining Notch activity and repressing NK-cell fate. Nat. Commun. 2016, 7, 11171. [Google Scholar] [CrossRef] [PubMed]
- Del Real, M.M.; Rothenberg, E.V. Architecture of a lymphomyeloid developmental switch controlled by PU.1, Notch and GATA3. Development 2013, 140, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chan, H.L.; Bai, F.; Ma, J.; Scott, A.; Robbins, D.J.; Capobianco, A.J.; Zhu, P.; Pei, X.H. GATA3 restrains B cell proliferation and cooperates with p18INK4c to repress B cell lymphomagenesis. Oncotarget 2016, 7, 64007–64020. [Google Scholar] [PubMed]
- Shaknovich, R.; Melnick, A. Epigenetics and B-cell Lymphoma. Curr. Opin. Hematol. 2011, 18, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kueh, H.Y.; Champhekar, A.; Nutt, S.L.; Elowitz, M.B.; Rothenberg, E.V. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 2013, 341, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, L.; Klemsz, M.J. DNA methylation and chromatin structure regulate PU.1 expression. DNA Cell Biol. 1999, 18, 875–884. [Google Scholar] [CrossRef] [PubMed]
- De la Rica, L.; Rodríguez-Ubreva, J.; García, M.; Islam, A.B.; Urquiza, J.M.; Hernando, H.; Christensen, J.; Helin, K.; Gómez-Vaquero, C.; Ballestar, E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013, 14, R99. [Google Scholar] [CrossRef] [PubMed]
- Tatetsu, H.; Ueno, S.; Hata, H.; Yamada, Y.; Takeya, M.; Mitsuya, H.; Tenen, D.G.; Okuno, Y. Down-regulation of PU.1 by methylation of distal regulatory elements and the promoter is required for myeloma cell growth. Cancer Res. 2007, 67, 5328–5336. [Google Scholar] [CrossRef] [PubMed]
- Sonnet, M.; Claus, R.; Becker, N.; Zucknick, M.; Petersen, J.; Lipka, D.B.; Oakes, C.C.; Andrulis, M.; Lier, A.; Milsom, M.D.; et al. Early aberrant DNA methylation events in a mouse model of acute myeloid leukemia. Genome Med. 2014, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Schnerch, D.; Yalcintepe, J.; Schmidts, A.; Becker, H.; Follo, M.; Engelhardt, M.; Wäsch, R. Cell cycle control in acute myeloid leukemia. Am. J. Cancer Res. 2012, 2, 508–528. [Google Scholar] [PubMed]
- Alon, U. Network motifs: Theory and experimental approaches. Nat. Rev. Genet. 2007, 8, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Grandy, R.A.; Whitfield, T.W.; Wu, H.; Fitzgerald, M.P.; van Oudenhove, J.J.; Zaidi, S.K.; Montecino, M.A.; Lian, J.B.; van Wijnen, A.J.; Stein, J.L.; et al. Genome-wide studies reveal that H3K4me3 modification in bivalent genes is dynamically regulated during the pluripotent cell cycle and stabilized upon differentiation. Mol. Cell. Biol. 2015, 36, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Dodd, I.B.; Micheelsen, M.A.; Sneppen, K.; Thon, G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell 2007, 129, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Sneppen, K.; Dodd, I.B. A simple histone code opens many paths to epigenetics. PLoS Comput. Biol. 2012, 8, e1002643. [Google Scholar] [CrossRef] [PubMed]
- Anink-Groenen, L.C.; Maarleveld, T.R.; Verschure, P.J.; Bruggeman, F.J. Mechanistic stochastic model of histone modification pattern formation. Epigenet. Chromatin 2014, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Ku, W.L.; Girvan, M.; Yuan, G.C.; Sorrentino, F.; Ott, E. Modeling the dynamics of bivalent histone modifications. PLoS ONE 2013, 8, e77944. [Google Scholar] [CrossRef] [PubMed]
- Bröske, A.M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Rosenbauer, F.; Wagner, K.; Kutok, J.L.; Iwasaki, H.; Le Beau, M.M.; Okuno, Y.; Akashi, K.; Fiering, S.; Tenen, D.G. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 2004, 36, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG island hypermethylation mediated by DNMT3A is a consequence of AML Progression. Cell 2017, 168, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Wirth, H.; Galle, J. Gene expression density profiles characterize modes of genomic regulation: Theory and experiment. J. Biotechnol. 2010, 149, 98–114. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thalheim, T.; Herberg, M.; Loeffler, M.; Galle, J. The Regulatory Capacity of Bivalent Genes—A Theoretical Approach. Int. J. Mol. Sci. 2017, 18, 1069. https://doi.org/10.3390/ijms18051069
Thalheim T, Herberg M, Loeffler M, Galle J. The Regulatory Capacity of Bivalent Genes—A Theoretical Approach. International Journal of Molecular Sciences. 2017; 18(5):1069. https://doi.org/10.3390/ijms18051069
Chicago/Turabian StyleThalheim, Torsten, Maria Herberg, Markus Loeffler, and Joerg Galle. 2017. "The Regulatory Capacity of Bivalent Genes—A Theoretical Approach" International Journal of Molecular Sciences 18, no. 5: 1069. https://doi.org/10.3390/ijms18051069
APA StyleThalheim, T., Herberg, M., Loeffler, M., & Galle, J. (2017). The Regulatory Capacity of Bivalent Genes—A Theoretical Approach. International Journal of Molecular Sciences, 18(5), 1069. https://doi.org/10.3390/ijms18051069