Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies

Abstract
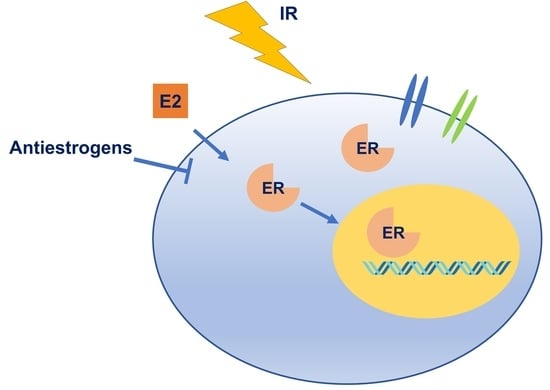
:
1. Introduction
2. Estrogen Receptor Signaling and Ionizing Radiation
2.1. Molecular and Cellular Responses to Ionizing Radiation
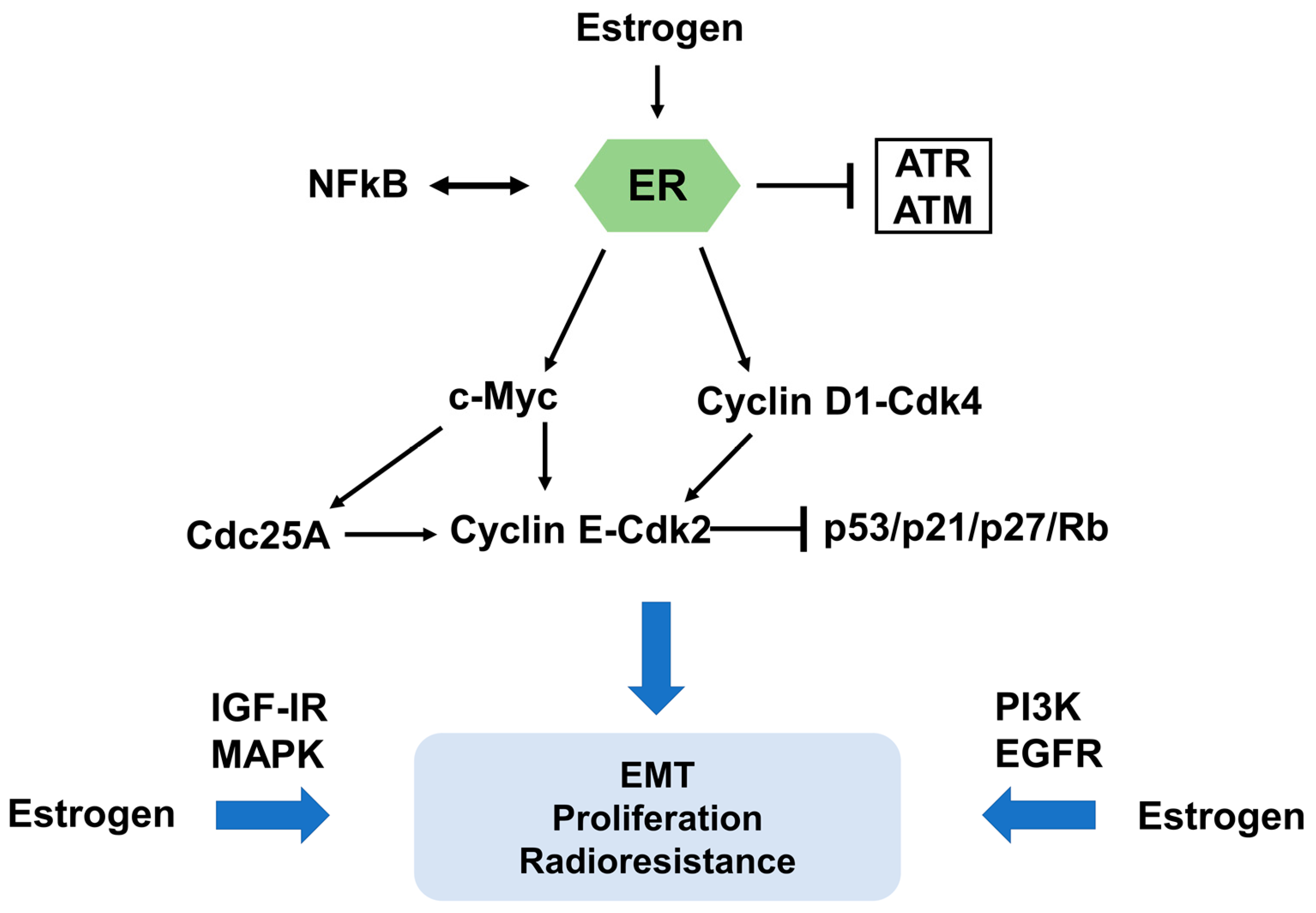
2.2. Interaction between Estrogen Receptor Signaling and Ionizing Irradiation
3. The Combination of Anti-Estrogen and Irradiation Therapy in Cancer
4. ER Signaling in Head and Neck Cancer
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gustafsson, J.A. The different roles of er subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Zhao, H.; Fuqua, S.A. Selective estrogen receptor modulators: Structure, function, and clinical use. J. Clin. Oncol. 2000, 18, 3172–3186. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R.; Fuqua, S.A.; Shou, J. Estrogen receptor: Current understanding of its activation and modulation. Clin. Cancer Res. 2001, 7, 4338s–4342s. [Google Scholar] [PubMed]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Korach, K.S. Estrogen receptors and human disease: An update. Arch. Toxicol. 2012, 86, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Ellem, S.J.; Risbridger, G.P. Treating prostate cancer: A rationale for targeting local oestrogens. Nat. Rev. Cancer 2007, 7, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, S.J. DNA damage by drugs and radiation: What is important and how is it measured? Eur. J. Cancer 1992, 28, 273–276. [Google Scholar] [CrossRef]
- Ward, J.F. DNA damage as the cause of ionizing radiation-induced gene activation. Radiat. Res. 1994, 138, S85–S88. [Google Scholar] [CrossRef] [PubMed]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal. 2014, 21, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Leadon, S.A. Repair of DNA damage produced by ionizing radiation: A minireview. Semin. Radiat. Oncol. 1996, 6, 295–305. [Google Scholar] [CrossRef]
- Borrego-Soto, G.; Ortiz-Lopez, R.; Rojas-Martinez, A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet. Mol. Biol. 2015, 38, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.W.; Miller, A.C. Effects of scavengers of reactive oxygen and radical species on cell survival following photodynamic treatment in vitro: Comparison to ionizing radiation. Radiat. Res. 1986, 108, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.; Deoraj, A.; Roy, D. Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim. Biophys. Acta 2011, 1815, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Kasparek, T.R.; Humphrey, T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011, 22, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Pallis, A.G.; Karamouzis, M.V. DNA repair pathways and their implication in cancer treatment. Cancer Metastasis Rev. 2010, 29, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Malu, S.; Malshetty, V.; Francis, D.; Cortes, P. Role of non-homologous end joining in V(D)J recombination. Immunol. Res. 2012, 54, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Wu, J.; Baloue, K.K.; Crowe, D.L. Regulation of the nijmegen breakage syndrome 1 gene NBS1 by c-Myc, p53 and coactivators mediates estrogen protection from DNA damage in breast cancer cells. Int. J. Oncol. 2013, 42, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Xia, F.; Mahalingam, M.; Virbasius, C.M.; Wajapeyee, N.; Green, M.R. MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Mol. Cell. Biol. 2013, 33, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, K.; Pannizzo, P.; Lassak, A.; Gualco, E.; Surmacz, E.; Croul, S.; Del Valle, L.; Khalili, K.; Reiss, K. Estrogen receptor β-mediated nuclear interaction between IRS-1 and Rad51 inhibits homologous recombination directed DNA repair in medulloblastoma. J. Cell. Physiol. 2009, 219, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. Linking DNA damage and hormone signaling pathways in cancer. Trends Endocrinol. Metab. 2016, 27, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Sausville, E.A. Drug discovery targeting Chk1 and Chk2 kinases. Prog. Cell Cycle Res. 2003, 5, 413–421. [Google Scholar] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Krempler, A.; Deckbar, D.; Jeggo, P.A.; Lobrich, M. An imperfect G2M checkpoint contributes to chromosome instability following irradiation of s and G2 phase cells. Cell Cycle 2007, 6, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Feigelson, H.S.; Ross, R.K.; Yu, M.C.; Coetzee, G.A.; Reichardt, J.K.; Henderson, B.E. Genetic susceptibility to cancer from exogenous and endogenous exposures. J. Cell. Biochem. Suppl. 1996, 25, 15–22. [Google Scholar] [CrossRef]
- Foster, J.S.; Henley, D.C.; Bukovsky, A.; Seth, P.; Wimalasena, J. Multifaceted regulation of cell cycle progression by estrogen: Regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function. Mol. Cell. Biol. 2001, 21, 794–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Wu, S.P.; Liu, J.B.; Shi, Y.S.; Huang, X.; Zhang, Q.B.; Yao, K.T. Myc regulation of Chk1 and Chk2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Cole, M.D. Turning the tables: Myc activates wnt in breast cancer. Cell Cycle 2007, 6, 2625–2627. [Google Scholar] [CrossRef] [PubMed]
- Dubik, D.; Shiu, R.P. Mechanism of estrogen activation of c-Myc oncogene expression. Oncogene 1992, 7, 1587–1594. [Google Scholar] [PubMed]
- Watson, P.H.; Pon, R.T.; Shiu, R.P. Inhibition of c-Myc expression by phosphorothioate antisense oligonucleotide identifies a critical role for c-Myc in the growth of human breast cancer. Cancer Res. 1991, 51, 3996–4000. [Google Scholar] [PubMed]
- Prall, O.W.; Rogan, E.M.; Sutherland, R.L. Estrogen regulation of cell cycle progression in breast cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 65, 169–174. [Google Scholar] [CrossRef]
- Watts, C.K.; Brady, A.; Sarcevic, B.; deFazio, A.; Musgrove, E.A.; Sutherland, R.L. Antiestrogen inhibition of cell cycle progression in breast cancer cells in associated with inhibition of cyclin-dependent kinase activity and decreased retinoblastoma protein phosphorylation. Mol. Endocrinol. 1995, 9, 1804–1813. [Google Scholar] [PubMed]
- Altucci, L.; Addeo, R.; Cicatiello, L.; Dauvois, S.; Parker, M.G.; Truss, M.; Beato, M.; Sica, V.; Bresciani, F.; Weisz, A. 17β-estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G1-arrested human breast cancer cells. Oncogene 1996, 12, 2315–2324. [Google Scholar] [PubMed]
- Sabbah, M.; Courilleau, D.; Mester, J.; Redeuilh, G. Estrogen induction of the cyclin D1 promoter: Involvement of a camp response-like element. Proc. Natl. Acad. Sci. USA 1999, 96, 11217–11222. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rivera, E.; Samudio, I.; Safe, S. Estrogen regulation of cyclin D1 gene expression in ZR-75 breast cancer cells involves multiple enhancer elements. J. Biol. Chem. 2001, 276, 30853–30861. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, N.R.; Prall, O.W.; Musgrove, E.A.; Sutherland, R.L. Inducible overexpression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of antiestrogens. Clin. Cancer Res. 1997, 3, 849–854. [Google Scholar] [PubMed]
- Toillon, R.A.; Magne, N.; Laios, I.; Lacroix, M.; Duvillier, H.; Lagneaux, L.; Devriendt, D.; Van Houtte, P.; Leclercq, G. Interaction between estrogen receptor α, ionizing radiation and (anti-) estrogens in breast cancer cells. Breast Cancer Res. Treat. 2005, 93, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Toillon, R.A.; Magne, N.; Laios, I.; Castadot, P.; Kinnaert, E.; Van Houtte, P.; Desmedt, C.; Leclercq, G.; Lacroix, M. Estrogens decrease γ-ray-induced senescence and maintain cell cycle progression in breast cancer cells independently of p53. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Molinari, A.M.; Bontempo, P.; Schiavone, E.M.; Tortora, V.; Verdicchio, M.A.; Napolitano, M.; Nola, E.; Moncharmont, B.; Medici, N.; Nigro, V.; et al. Estradiol induces functional inactivation of p53 by intracellular redistribution. Cancer Res. 2000, 60, 2594–2597. [Google Scholar] [PubMed]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef] [PubMed]
- Magne, N.; Toillon, R.A.; Bottero, V.; Didelot, C.; Houtte, P.V.; Gerard, J.P.; Peyron, J.F. NFκB modulation and ionizing radiation: Mechanisms and future directions for cancer treatment. Cancer Lett. 2006, 231, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; Van Dam, P.; Pauwels, P.; Dirix, L.Y.; Van Laere, S.J. The interaction between er and NFκB in resistance to endocrine therapy. Breast Cancer Res. BCR 2012, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Egloff, A.M.; Rothstein, M.E.; Seethala, R.; Siegfried, J.M.; Grandis, J.R.; Stabile, L.P. Cross-talk between estrogen receptor and epidermal growth factor receptor in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, J.M.; Hershberger, P.A.; Stabile, L.P. Estrogen receptor signaling in lung cancer. Semin. Oncol. 2009, 36, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.H.; Chu, N.M.; Kao, S.H. Estrogen, estrogen receptor and lung cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Mawson, A.; Lai, A.; Carroll, J.S.; Sergio, C.M.; Mitchell, C.J.; Sarcevic, B. Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer cells through differential regulation of c-Myc and cyclin D1. Mol. Cell. Endocrinol. 2005, 229, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Garban, D.C.; Chen, H.W.; Fishbein, M.C.; Goodglick, L.; Pietras, R.J. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids 2007, 72, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Liu, Y.; Zhou, L.J.; Wang, Z.Q.; Wu, Z.H.; Yang, X.Y. Role of estrogen in lung cancer based on the estrogen receptor-epithelial mesenchymal transduction signaling pathways. OncoTargets Ther. 2015, 8, 2849–2863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Nie, Y.; Lv, M.; He, L.; Wang, T.; Hou, Y. ERβ-mediated estradiol enhances epithelial mesenchymal transition of lung adenocarcinoma through increasing transcription of midkine. Mol. Endocrinol. 2012, 26, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; La Fleur, L.; Melissaridou, S.; Roberg, K. The relationship between EMT, CD44(high) /EGFR(low) phenotype, and treatment response in head and neck cancer cell lines. J. Oral Pathol. Med. 2016, 45, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Jutten, B.; Habets, R.; Paesmans, K.; Groot, A.J.; Lambin, P.; Wouters, B.G.; Lammering, G.; Vooijs, M. E-cadherin loss associated with emt promotes radioresistance in human tumor cells. Radiother. Oncol. 2011, 99, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 4, e875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, H.; Jiang, Z.; Yue, J.; Hou, Q.; Xie, R.; Wu, S. Fractionated irradiation-induced EMT-like phenotype conferred radioresistance in esophageal squamous cell carcinoma. J. Radiat. Res. 2016, 57, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.P.; Jones, C.T.; Todd, I.D. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br. J. Cancer 1971, 25, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Wazer, D.E.; Tercilla, O.F.; Lin, P.S.; Schmidt-Ullrich, R. Modulation in the radiosensitivity of MCF-7 human breast carcinoma cells by 17β-estradiol and tamoxifen. Br. J. Radiol. 1989, 62, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Wazer, D.E.; Joyce, M.; Jung, L.; Band, V. Alterations in growth phenotype and radiosensitivity after fractionated irradiation of breast carcinoma cells from a single patient. Int. J. Radiat. Oncol. Biol. Phys. 1993, 26, 81–88. [Google Scholar] [CrossRef]
- Villalobos, M.; Aranda, M.; Nunez, M.I.; Becerra, D.; Olea, N.; Ruiz de Almodovar, M.; Pedraza, V. Interaction between ionizing radiation, estrogens and antiestrogens in the modification of tumor microenvironment in estrogen dependent multicellular spheroids. Acta Oncol. 1995, 34, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, M.; Becerra, D.; Nunez, M.I.; Valenzuela, M.T.; Siles, E.; Olea, N.; Pedraza, V.; Ruiz de Almodovar, J.M. Radiosensitivity of human breast cancer cell lines of different hormonal responsiveness. Modulatory effects of oestradiol. Int. J. Radiat. Biol. 1996, 70, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, G.H.; Strickert, T.; Marthinsen, A.B.; Lundgren, S. Changes in radiation sensitivity and steroid receptor content induced by hormonal agents and ionizing radiation in breast cancer cells in vitro. Acta Oncol. 1996, 35, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.J.; Schlatterer, K.; Stalla, G.K.; Von Angerer, E.; Wowra, B. Pharmacological enhancement of radiosurgery response: Studies on an in vitro model system. J. Radiosurg. 1998, 1, 51–56. [Google Scholar] [CrossRef]
- Yang, L.; Yuan, X.; Wang, J.; Gu, C.; Zhang, H.; Yu, J.; Liu, F. Radiosensitization of human glioma cells by tamoxifen is associated with the inhibition of PKC-I activity in vitro. Oncol. lett. 2015, 10, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Q.; Haffty, B.G.; Li, X.; Moran, M.S. Fulvestrant radiosensitizes human estrogen receptor-positive breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 431, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Miller, E.M.; Parker, C.J.; Jordan, V.C.; Mulcahy, R.T. 4-hydroxytamoxifen, an active metabolite of tamoxifen, does not alter the radiation sensitivity of MCF-7 breast carcinoma cells irradiated in vitro. Breast Cancer Res. Treat. 1994, 30, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, M.M.; Hyland, E.; Campiani, G.; Ramunno, A.; Nacci, V.; Zisterer, D.M. Caspase-3 is not essential for DNA fragmentation in MCF-7 cells during apoptosis induced by the pyrrolo-1,5-benzoxazepine, pbox-6. FEBS Lett. 2002, 515, 66–70. [Google Scholar] [CrossRef]
- Kantorowitz, D.A.; Thompson, H.J.; Furmanski, P. Effect of conjoint administration of tamoxifen and high-dose radiation on the development of mammary carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 1993, 26, 89–94. [Google Scholar] [CrossRef]
- Inano, H.; Onoda, M. Prevention of radiation-induced mammary tumors. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 212–223. [Google Scholar] [CrossRef]
- Inano, H.; Onoda, M.; Suzuki, K.; Kobayashi, H.; Wakabayashi, K. Prevention of radiation-induced mammary tumours in rats by combined use of WR-2721 and tamoxifen. Int. J. Radiat. Biol. 2000, 76, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Mendoza, M.; Jurado, R.; Mir, R.; Medina, L.A.; Prado-Garcia, H.; Garcia-Lopez, P. Antihormonal agents as a strategy to improve the effect of chemo-radiation in cervical cancer: In vitro and in vivo study. BMC Cancer 2015, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Azria, D.; Larbouret, C.; Cunat, S.; Ozsahin, M.; Gourgou, S.; Martineau, P.; Evans, D.B.; Romieu, G.; Pujol, P.; Pelegrin, A. Letrozole sensitizes breast cancer cells to ionizing radiation. Breast Cancer Res. BCR 2005, 7, R156–R163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anscher, M.S.; Kong, F.M.; Andrews, K.; Clough, R.; Marks, L.B.; Bentel, G.; Jirtle, R.L. Plasma transforming growth factor β1 as a predictor of radiation pneumonitis. Int. J. Radiat. Oncol. Biol. Phys. 1998, 41, 1029–1035. [Google Scholar] [CrossRef]
- Anscher, M.S.; Kong, F.M.; Jirtle, R.L. The relevance of transforming growth factor β 1 in pulmonary injury after radiation therapy. Lung Cancer 1998, 19, 109–120. [Google Scholar] [CrossRef]
- Chen, Y.; Williams, J.; Ding, I.; Hernady, E.; Liu, W.; Smudzin, T.; Finkelstein, J.N.; Rubin, P.; Okunieff, P. Radiation pneumonitis and early circulatory cytokine markers. Semin. Radiat. Oncol. 2002, 12, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Butta, A.; MacLennan, K.; Flanders, K.C.; Sacks, N.P.; Smith, I.; McKinna, A.; Dowsett, M.; Wakefield, L.M.; Sporn, M.B.; Baum, M.; et al. Induction of transforming growth factor β 1 in human breast cancer in vivo following tamoxifen treatment. Cancer Res. 1992, 52, 4261–4264. [Google Scholar] [PubMed]
- Dalberg, K.; Johansson, H.; Johansson, U.; Rutqvist, L.E. A randomized trial of long term adjuvant tamoxifen plus postoperative radiation therapy versus radiation therapy alone for patients with early stage breast carcinoma treated with breast-conserving surgery. Stockholm breast cancer study group. Cancer 1998, 82, 2204–2211. [Google Scholar] [CrossRef]
- Fisher, B.; Dignam, J.; Wolmark, N.; Wickerham, D.L.; Fisher, E.R.; Mamounas, E.; Smith, R.; Begovic, M.; Dimitrov, N.V.; Margolese, R.G.; et al. Tamoxifen in treatment of intraductal breast cancer: National surgical adjuvant breast and bowel project B-24 randomised controlled trial. Lancet 1999, 353, 1993–2000. [Google Scholar] [CrossRef]
- Ahn, P.H.; Vu, H.T.; Lannin, D.; Obedian, E.; DiGiovanna, M.P.; Burtness, B.; Haffty, B.G. Sequence of radiotherapy with tamoxifen in conservatively managed breast cancer does not affect local relapse rates. J. Clin. Oncol. 2005, 23, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.E.; Christensen, V.J.; Hwang, W.T.; Fox, K.; Solin, L.J. Impact of concurrent versus sequential tamoxifen with radiation therapy in early-stage breast cancer patients undergoing breast conservation treatment. J. Clin. Oncol. 2005, 23, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Pierce, L.J.; Hutchins, L.F.; Green, S.R.; Lew, D.L.; Gralow, J.R.; Livingston, R.B.; Osborne, C.K.; Albain, K.S. Sequencing of tamoxifen and radiotherapy after breast-conserving surgery in early-stage breast cancer. J. Clin. Oncol. 2005, 23, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Ishitobi, M.; Komoike, Y.; Motomura, K.; Koyama, H.; Nishiyama, K.; Inaji, H. Retrospective analysis of concurrent vs. Sequential administration of radiotherapy and hormone therapy using aromatase inhibitor for hormone receptor-positive postmenopausal breast cancer. Anticancer Res. 2009, 29, 4791–4794. [Google Scholar] [PubMed]
- Ishitobi, M.; Shiba, M.; Nakayama, T.; Motomura, K.; Koyama, H.; Nishiyama, K.; Tamaki, Y. Treatment sequence of aromatase inhibitors and radiotherapy and long-term outcomes of breast cancer patients. Anticancer Res. 2014, 34, 4311–4314. [Google Scholar] [PubMed]
- Valakh, V.; Trombetta, M.G.; Werts, E.D.; Labban, G.; Khalid, M.K.; Kaminsky, A.; Parda, D. Influence of concurrent anastrozole on acute and late side effects of whole breast radiotherapy. Am. J. Clin. Oncol. 2011, 34, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Azria, D.; Belkacemi, Y.; Romieu, G.; Gourgou, S.; Gutowski, M.; Zaman, K.; Moscardo, C.L.; Lemanski, C.; Coelho, M.; Rosenstein, B.; et al. Concurrent or sequential adjuvant letrozole and radiotherapy after conservative surgery for early-stage breast cancer (CO-HO-RT): A phase 2 randomised trial. Lancet Oncol. 2010, 11, 258–265. [Google Scholar] [CrossRef]
- Whelan, T.; Levine, M. Radiation therapy and tamoxifen: Concurrent or sequential? That is the question. J. Clin. Oncol. 2005, 23, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Chargari, C.; Toillon, R.A.; Macdermed, D.; Castadot, P.; Magne, N. Concurrent hormone and radiation therapy in patients with breast cancer: What is the rationale? Lancet Oncol. 2009, 10, 53–60. [Google Scholar] [CrossRef]
- Varga, Z.; Cserhati, A.; Kelemen, G.; Boda, K.; Thurzo, L.; Kahan, Z. Role of systemic therapy in the development of lung sequelae after conformal radiotherapy in breast cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Koc, M.; Polat, P.; Suma, S. Effects of tamoxifen on pulmonary fibrosis after cobalt-60 radiotherapy in breast cancer patients. Radiother. Oncol. 2002, 64, 171–175. [Google Scholar] [CrossRef]
- Bentzen, S.M.; Skoczylas, J.Z.; Overgaard, M.; Overgaard, J. Radiotherapy-related lung fibrosis enhanced by tamoxifen. J. Natl. Cancer Inst. 1996, 88, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Fowble, B.; Fein, D.A.; Hanlon, A.L.; Eisenberg, B.L.; Hoffman, J.P.; Sigurdson, E.R.; Daly, M.B.; Goldstein, L.J. The impact of tamoxifen on breast recurrence, cosmesis, complications, and survival in estrogen receptor-positive early-stage breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 669–677. [Google Scholar] [CrossRef]
- Dahhan, T.; Fons, G.; Buist, M.R.; Ten Kate, F.J.; van der Velden, J. The efficacy of hormonal treatment for residual or recurrent low-grade endometrial stromal sarcoma. A retrospective study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Grünow, J.; Rong, C.; Hischmann, J.; Zaoui, K.; Flechtenmacher, C.; Weber, K.J.; Plinkert, P.; Hess, J. Regulation of submaxillary gland androgen-regulated protein 3A via estrogen receptor 2 in radioresistant head and neck squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2017, 36, 25. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Globocan 2012 v1.0, Cancer Incidence and Mortality Worldwide: Iarc Cancerbase No. 11. International Agency for Research on Cancer: Lyon, France. Available online: http://globocan.iarc.fr (accessed on 12 November 2017).
- Vineis, P.; Alavanja, M.; Buffler, P.; Fontham, E.; Franceschi, S.; Gao, Y.T.; Gupta, P.C.; Hackshaw, A.; Matos, E.; Samet, J.; et al. Tobacco and cancer: Recent epidemiological evidence. J. Natl. Cancer Inst. 2004, 96, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Valimaa, H.; Savolainen, S.; Soukka, T.; Silvoniemi, P.; Makela, S.; Kujari, H.; Gustafsson, J.A.; Laine, M. Estrogen receptor-β is the predominant estrogen receptor subtype in human oral epithelium and salivary glands. J. Endocrinol. 2004, 180, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.W.; Dobbins, T.A.; Tseung, J.; Tran, N.; Lee, C.S.; O’Brien, C.J.; Clark, J.; Rose, B.R. Oestrogen receptor β expression in pleomorphic adenomas of the parotid gland. J. Clin. Pathol. 2009, 62, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Shatalova, E.G.; Klein-Szanto, A.J.; Devarajan, K.; Cukierman, E.; Clapper, M.L. Estrogen and cytochrome P450 1B1 contribute to both early- and late-stage head and neck carcinogenesis. Cancer Prev. Res. (Phila.) 2011, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Sumida, T.; Ishikawa, A.; Mori, Y. Stimulation of the estrogen axis induces epithelial-mesenchymal transition in human salivary cancer cells. Cancer Genom. Proteom. 2016, 13, 305–310. [Google Scholar]
- Somers, K.D.; Koenig, M.; Schechter, G.L. Growth of head and neck squamous cell carcinoma in nude mice: Potentiation of laryngeal carcinoma by 17β-estradiol. J. Natl. Cancer Inst. 1988, 80, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Lukits, J.; Remenar, E.; Rásó, E.; Ladányi, A.; Kásler, M.; Tímár, J. Molecular identification, expression and prognostic role of estrogen- and progesterone receptors in head and neck cancer. Int. J. Oncol. 2007, 30, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Grsic, K.; Opacic, I.L.; Sitic, S.; Milkovic Perisa, M.; Suton, P.; Sarcevic, B. The prognostic significance of estrogen receptor β in head and neck squamous cell carcinoma. Oncol. Lett. 2016, 12, 3861–3865. [Google Scholar] [CrossRef] [PubMed]
- Fei, M.; Zhang, J.; Zhou, J.; Xu, Y.; Wang, J. Sex-related hormone receptor in laryngeal squamous cell carcinoma: Correlation with androgen estrogen-α and prolactin receptor expression and influence of prognosis. Acta Otolaryngol. 2018, 138, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, B.; Tang, Q.; Liu, J.; Yang, X. Bisphenol a triggers proliferation and migration of laryngeal squamous cell carcinoma via gper mediated upregulation of IL-6. Cell. Biochem. Funct. 2017, 35, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Brooks, Y.S.; Ostano, P.; Jo, S.H.; Dai, J.; Getsios, S.; Dziunycz, P.; Hofbauer, G.F.; Cerveny, K.; Chiorino, G.; Lefort, K.; et al. Multifactorial ERβ and NOTCH1 control of squamous differentiation and cancer. J. Clin. Investig. 2014, 124, 2260–2276. [Google Scholar] [CrossRef] [PubMed]




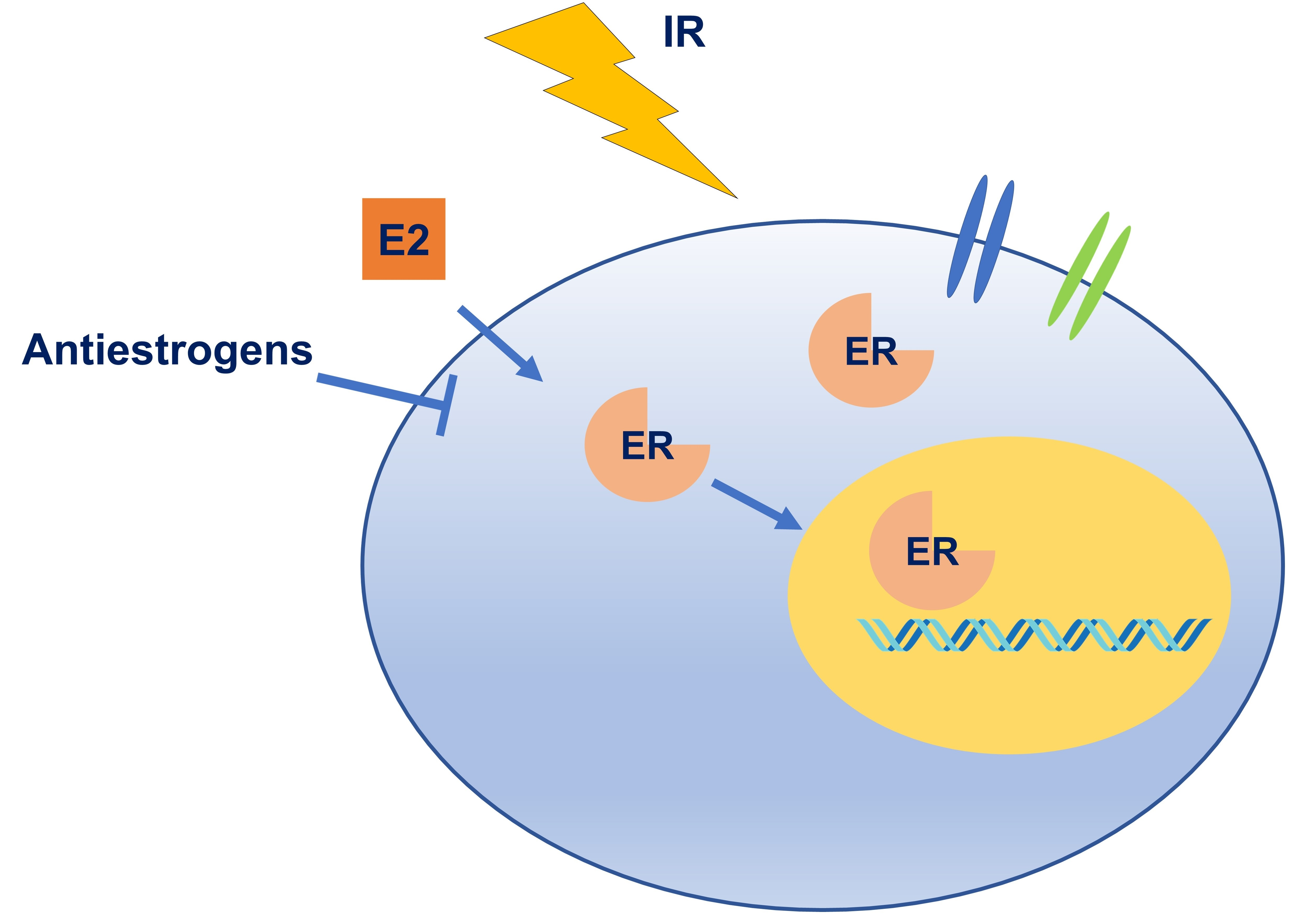{kind=link}
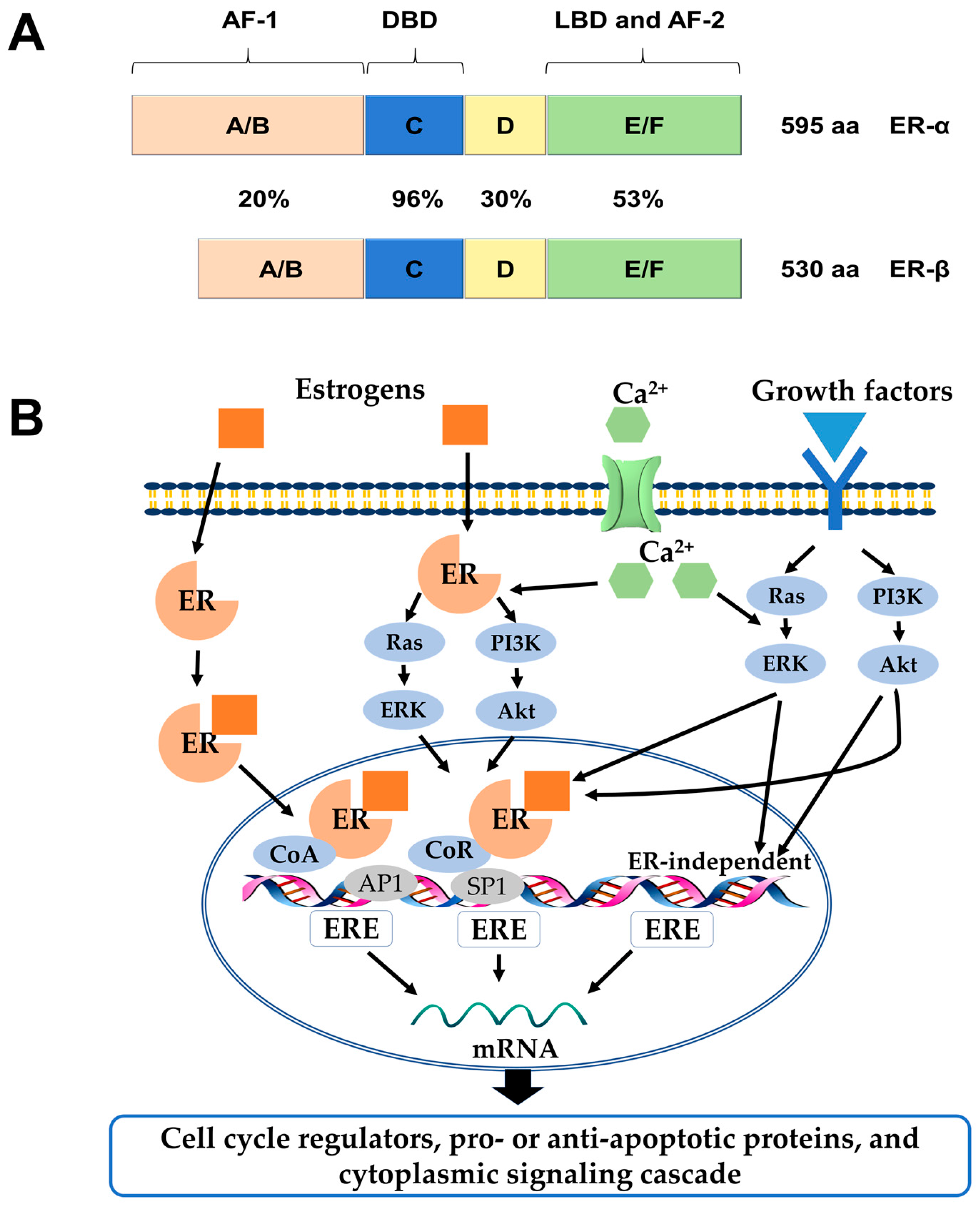{kind=link}
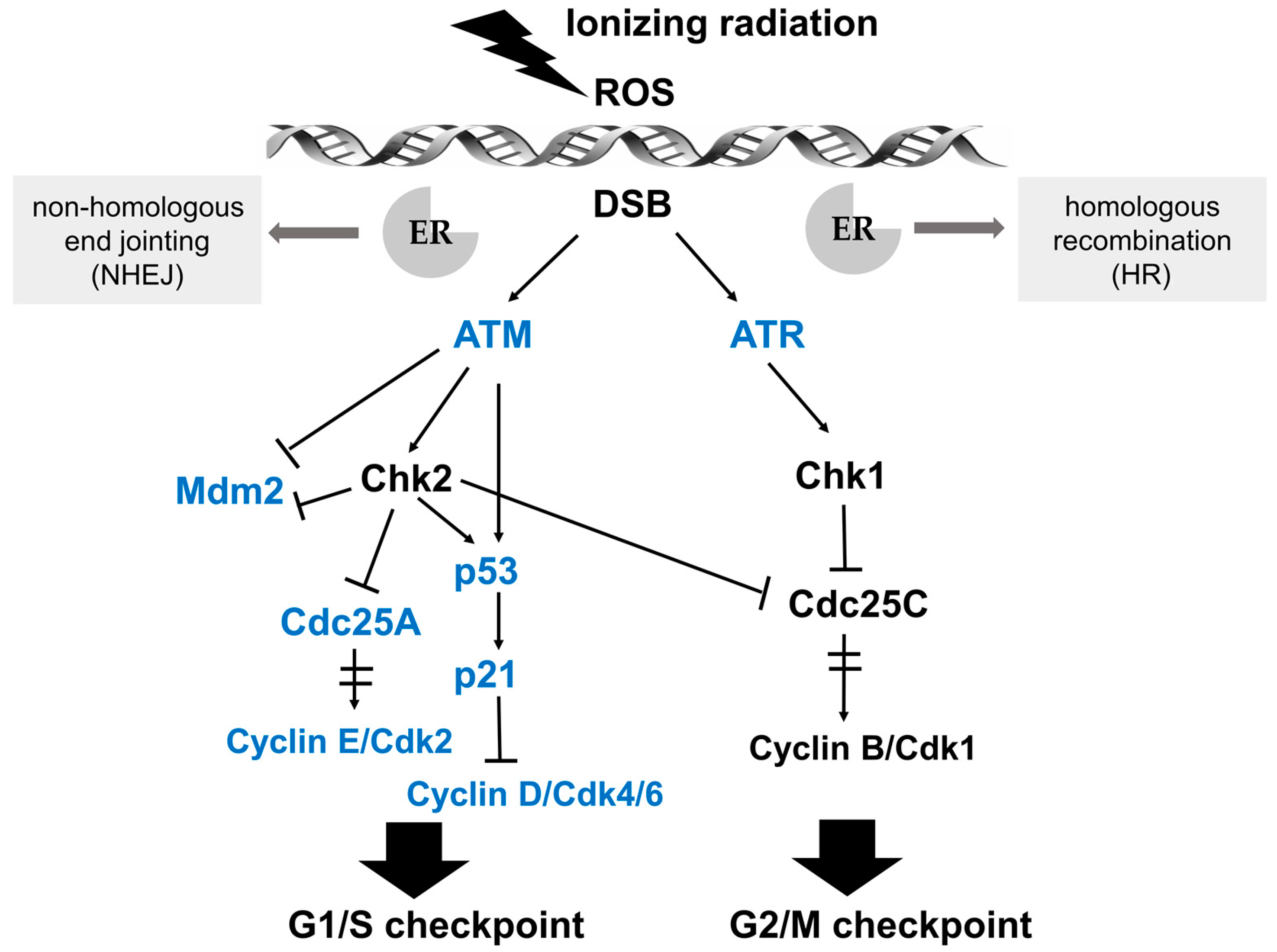{kind=link}
{kind=link}
| Type | Treatment Groups (n) | Tamoxifen or Aromatase Inhibitors | Radiotherapy | Chemotherapy (n) | Follow-up | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Retrospective 1976–1999 | Concurrent (254) vs. Sequential (241) | generally for 5 years | 48 Gy in 2 Gy Fractions with boost to primary tumor bed median total dose 64 Gy | CMF based (71) Adriamycin (42) other (16) none (371) | 10.4 years | No difference in overall survival (OS), HR, 1.234; 95% CI, 0.42 to 2.05; No difference in local recurrence, HR, 0.932; 95% CI 0.42 to 2.05 | [81] |
| Retrospective 1980–1995 | Concurrent (174) vs. Sequential (104) | 20 mg OD or 10 mg BID | Tangents only (182) or tangents and nodal (95) median total dose 64 Gy | Methotrexate-based (67) Doxorubicin-based (44) None (167) | 8.6 years | No difference in OS, HR 1.56; 95% CI, 0.87 to 2.79; No difference in relapse-free survival, HR 1.23; 95% CI, 0.63 to 2.41; No difference in local recurrence, HR 1.22; 95% CI, 0.33 to 4.49; No difference in cosmesis, or significant complications. | [82] |
| Retrospective 1989–1993 | Concurrent (202) vs. Sequential (107) | 20 mg daily for 5 years | 45–50 Gy to whole breast | cyclophosphamide, methotrexate, and fluorouracil (CMF) (156) cyclophosphamide, doxorubicin, and fluorouracil (CAF) (153) | 10.3 years | No difference in OS, HR 0.84; 95% CI 0.40 to 1.78; No difference in local recurrence, HR 0.73; 95% CI, 0.26 to 2.04; No difference in grade 3 or 4 hematologic toxicity. | [83] |
| Retrospective 2001–2008 | Concurrent (113) vs. Sequential (151) | anastrozole 1 mg or letrozole 2.5 mg daily for 5 years | 50 Gy in 2 Gy Fractions with boost to primary tumor bed median total dose 63.2 Gy | CMF (1) Taxane-based (7) Anthracycline-based (31) Combination of anthracycline and taxane (6) | 2.9 years | No differences in clinical outcome and treatment-related complications | [84] |
| Retrospective 2001–2009 | Concurrent (158) vs. Sequential (157) | anastrozole 1 mg or letrozole 2.5 mg daily for 5 years | 50 Gy in 2 Gy fractions with a boost of up to 63.2 Gy | Yes (57) None (258) | 5.6 years | No difference in disease-free survival. No difference in Grade 3 or 5 toxicities | [85] |
| Retrospective 1998–2008 | Concurrent (57) vs. Sequential (126) | Anastrozole or Tamoxifen | 45–54 Gy over an average of 49.5 days | anthracycline or taxane (51) none (132) | 2.3 years (Con) 2.6 years (Seq) | No difference in detectable breast fibrosis Concurrent (1.8%) vs. Sequential (4%) in Local recurrence | [86] |
| Randomized 2005–2007 | Concurrent (75) vs. Sequential (75) | 2.5 mg Letrozole daily for 5 years | A total dose of 50 Gy in 2 Gy fractions | FEC (28) None (122) | 2.2 years | No difference in subcutaneous fibrosis, lung fibrosis and quality of life | [87] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rong, C.; Meinert, É.F.R.C.; Hess, J. Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies. Int. J. Mol. Sci. 2018, 19, 713. https://doi.org/10.3390/ijms19030713
Rong C, Meinert ÉFRC, Hess J. Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies. International Journal of Molecular Sciences. 2018; 19(3):713. https://doi.org/10.3390/ijms19030713
Chicago/Turabian StyleRong, Chao, Étienne Fasolt Richard Corvin Meinert, and Jochen Hess. 2018. "Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies" International Journal of Molecular Sciences 19, no. 3: 713. https://doi.org/10.3390/ijms19030713
APA StyleRong, C., Meinert, É. F. R. C., & Hess, J. (2018). Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies. International Journal of Molecular Sciences, 19(3), 713. https://doi.org/10.3390/ijms19030713