1. Introduction
Normal physiological processes are maintained through a homeostatic balance between two very critical parts of the normal development and maturation cycle namely cell proliferation and cell death. Any alterations to this homeostatic balance can lead to diseases such as AIDS, diabetes, neurodegenerative diseases, and cancer.
Apoptosis is a cellular suicide mechanism that is regulated by a family of cysteine proteases otherwise known as caspases [
1]. Apoptosis is characterized by: a reduction in mitochondrial transmembrane potential, intracellular acidification, cell shrinkage, DNA fragmentation and condensation, production of reactive oxygen species, externalization of phosphatidylserine and selective proteolysis of a subset of cellular proteins [
2]. It is sub-classified into two non-exclusive types of death pathways namely the extrinsic (receptor-mediated) and intrinsic (mitochondria-mediated) pathway [
3]. Cancer cells have the ability to resist apoptotic insults by means of various mechanisms and therefore a thorough understanding of these mechanisms is imperative to unravel the secret to designing more effective and targeted therapeutic strategies [
4].
Anemone is a genus of more than 150 species of flowering plants that are native to the temperate zones of both the Northern and Southern hemispheres. Of the 150 species, more than 50 are used in various traditional medical systems. In China alone, 53 species, 9 subspecies, and 36 varieties are found in most provinces of which at least 38 species/varieties have ethnopharmacological uses [
5]. Observed pharmacological activities include anti-cancer, anti-microbial, anti-inflammatory, sedative, and analgesic activities as well as anti-convulsant and anti-histamine effects. Various parts of
A. nemorosa have been used for the treatment of numerous medical conditions such as headaches, tertian agues, rheumatic gout, leprosy, lethargy, eye inflammation, malignant ulcers and as an antimicrobial, antifungal, diuretic, and abortive agent [
5,
6].
Although
A. nemorosa is not used traditionally for anti-cancer treatment, reported evidence shows the presence of compounds responsible for anti-cancer activity such as triterpenoids and saponins [
7]. For this reason, we investigated the mode of cell death caused by an aqueous extract from
A. nemorosa (
Figure 1) on HeLa cervical cancer cells. The aqueous extract was selected based on a preliminary anti-proliferative screening of ethanolic, hydroethanolic, and aqueous extracts.
3. Discussion
To date there has been no evidence of the use of
A. nemorosa for the treatment of cancer. Studies involving
A. nemorosa mainly consist of anti-microbial activity against known hospital and fish pathogens such as
E. coli and
Vibrio anguillarum, respectively [
14,
15]. However, the isolation of numerous phytochemical compounds from other species of the
Anemone genus has provided sufficient reasons as to why
A. nemorosa could be used for cancer treatment.
Raddeanin A is a pentacyclic triterpenoid saponin, isolated from
A. raddeana, and induces apoptosis in multiple cell lines by means of increased Bax expression, reduced Bcl-2 and Survivin expression and the activation of caspases 3, 8 and 9 in gastric cancer cells [
5]. Whilst isolated triterpenoid saponins from
A. flaccida, hedera saponin StI4a, hedera saponin St-J, anhuienoside E, hedera saponin B and flaccidoside II, have been shown to induce apoptosis in HeLa cervical cancer cells [
7].
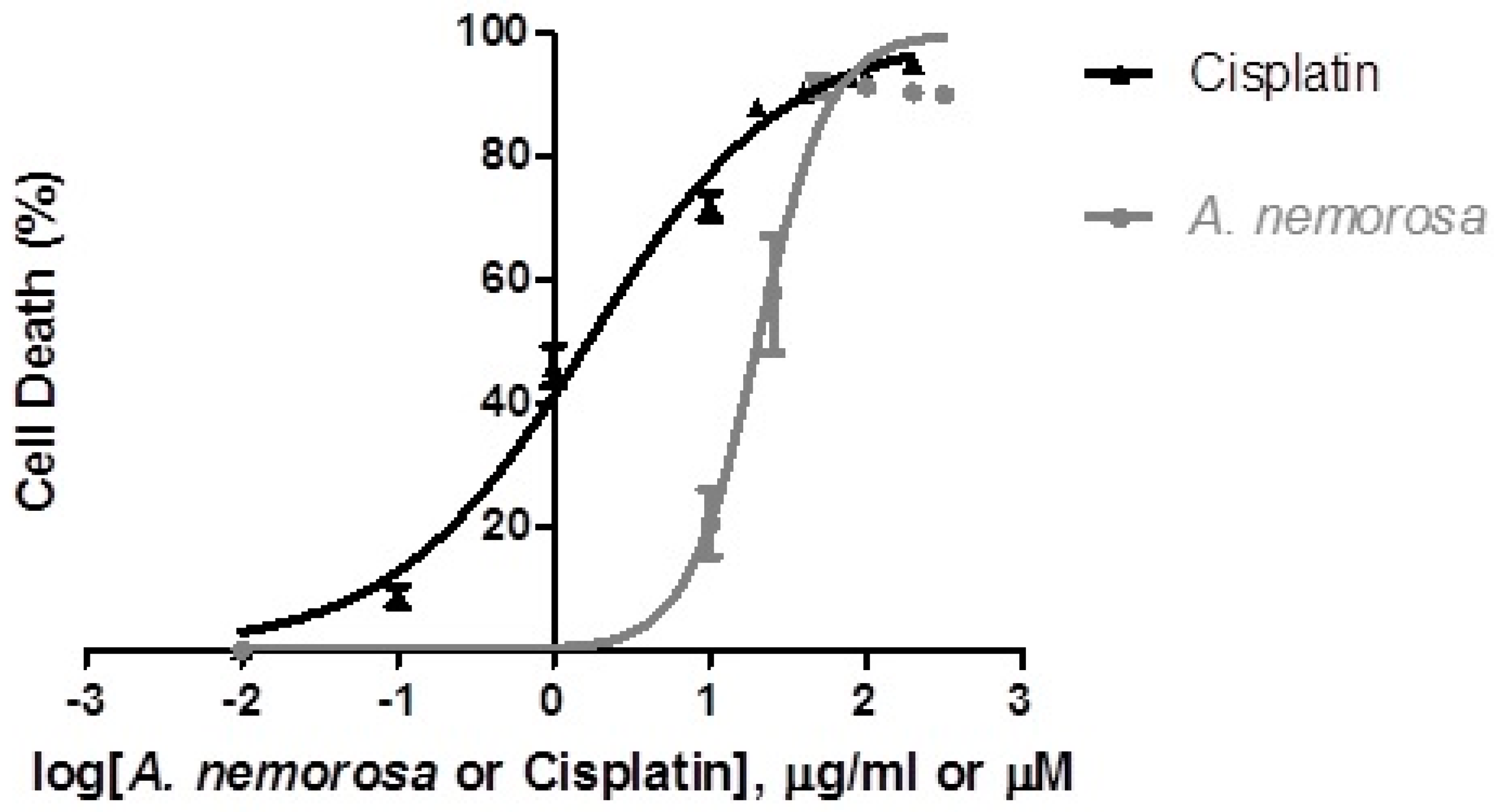
Dose-response assays (
Figure 1) were performed by means of the Hoechst 33342/PI dual staining method and results indicated that
A. nemorosa inhibits proliferation of HeLa cells in a dose dependent manner. For a crude extract to be considered promising for systemic application, the IC
50 should be less than 100 µg/mL in vitro [
16]. The IC
50 of
A. nemorosa was determined to be 20.33 ± 2.480 µg/mL.
One of the basic characteristics of cancer cells is their ability to proliferate uncontrollably. Therefore, DNA cell cycle analysis remains an important aspect in the search for new and improved chemotherapeutic agents. Uncontrollable proliferation can be ascribed to the alteration of the various cell cycle checkpoints [
17]. Extracts, such as
A. nemorosa, with cytotoxic activity may counteract these alterations by activating cell cycle arrest through the damage to the mitotic spindle or by affecting the signaling pathways that regulate proliferation [
18].
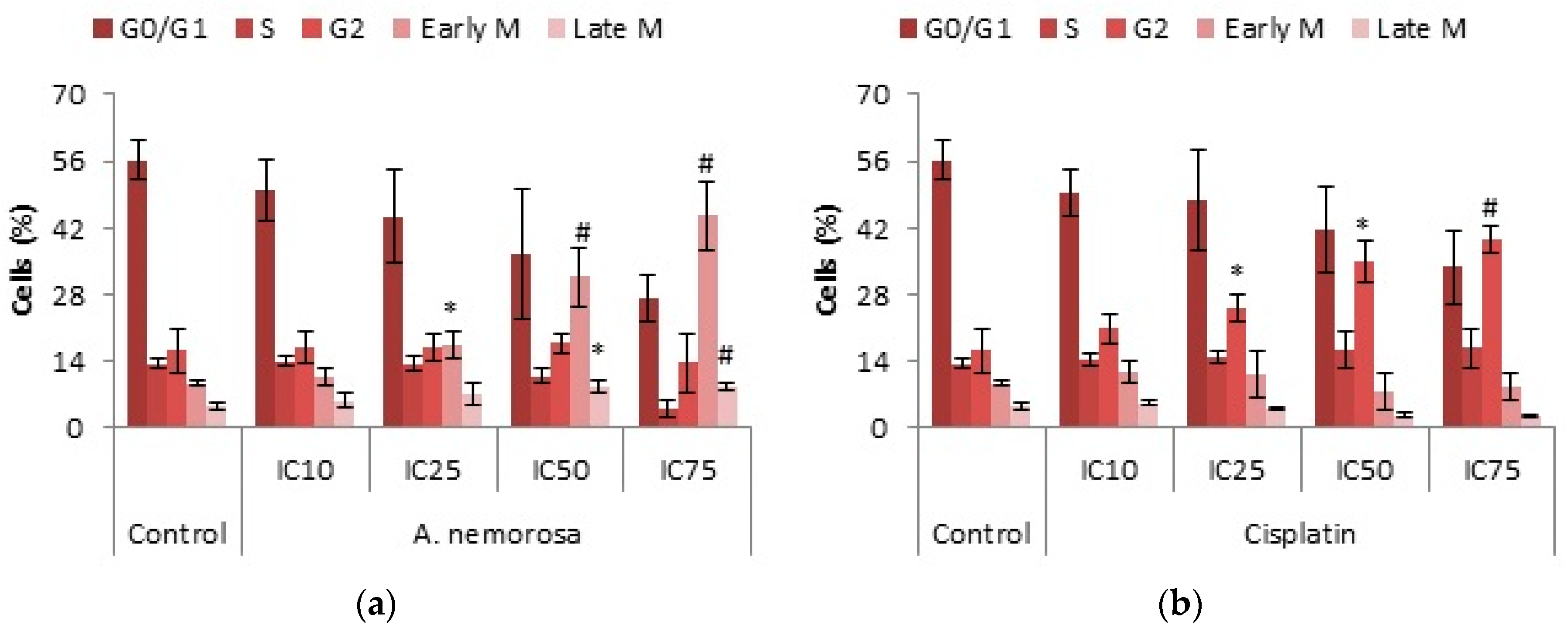
A. nemorosa was shown to dose dependently arrest HeLa cells in the early M phase of the cell cycle after 24 h of exposure whereas cisplatin arrested cells in the G2 phase (
Figure 3). The exact mechanism of how
A. nemorosa and cisplatin cause arrest in the early M and G2 phase, respectively, cannot be deduced from the NucRed staining and more than one possibility exists. There are two parallel cascades that ultimately serve to inactivate the CyclinB1-CDK1 complex subsequently blocking entry into mitosis. The first cascade occurs quite rapidly where Chk1 phosphorylates Cdc25C preventing it from activating CDK1, leading to G2 arrest. The slower second parallel cascade involves the phosphorylation of p53 by means of activating CKI’s (such as p21) which inactivate the CyclinB1-CDK1 complex. Mitotic arrest is mainly due to the disruption of the formation of the mitotic spindle resulting in mitotic catastrophe [
19].
Early M phase arrest by
A. nemorosa was confirmed through staining with phospho-H3 (ser10) antibody as the phosphorylation of histone H3 is believed to be a marker for cells entering into mitosis [
20]. Treatment with the IC
75 value of extract showed a significant increase in phosphorylated histone H3 after 24 h (
Table 2). Cisplatin treatments indicated a dose dependent decrease in phosphorylated histone H3 which supports the G2 phase arrest obtained during cell cycle analysis. Treatment with staurosporine has also showed to inhibit histone H3 phosphorylation leading to G2 phase arrest [
20]. Studies done by Hans and Dimitrov (2001) showed that during cell division, the phosphorylation of histone H3 is initiated at different phases. However, metaphase chromosomes have been found to always be the most heavily phosphorylatedand suggests a possible role for histone H3 in the passage of cells from metaphase to anaphase [
21]. The low level of phosphorylation seen in this study suggests that HeLa cells would then be mitotically arrested in metaphase which is relatively early in the process of mitosis supporting the early M phase arrest obtained for the cell cycle analysis.
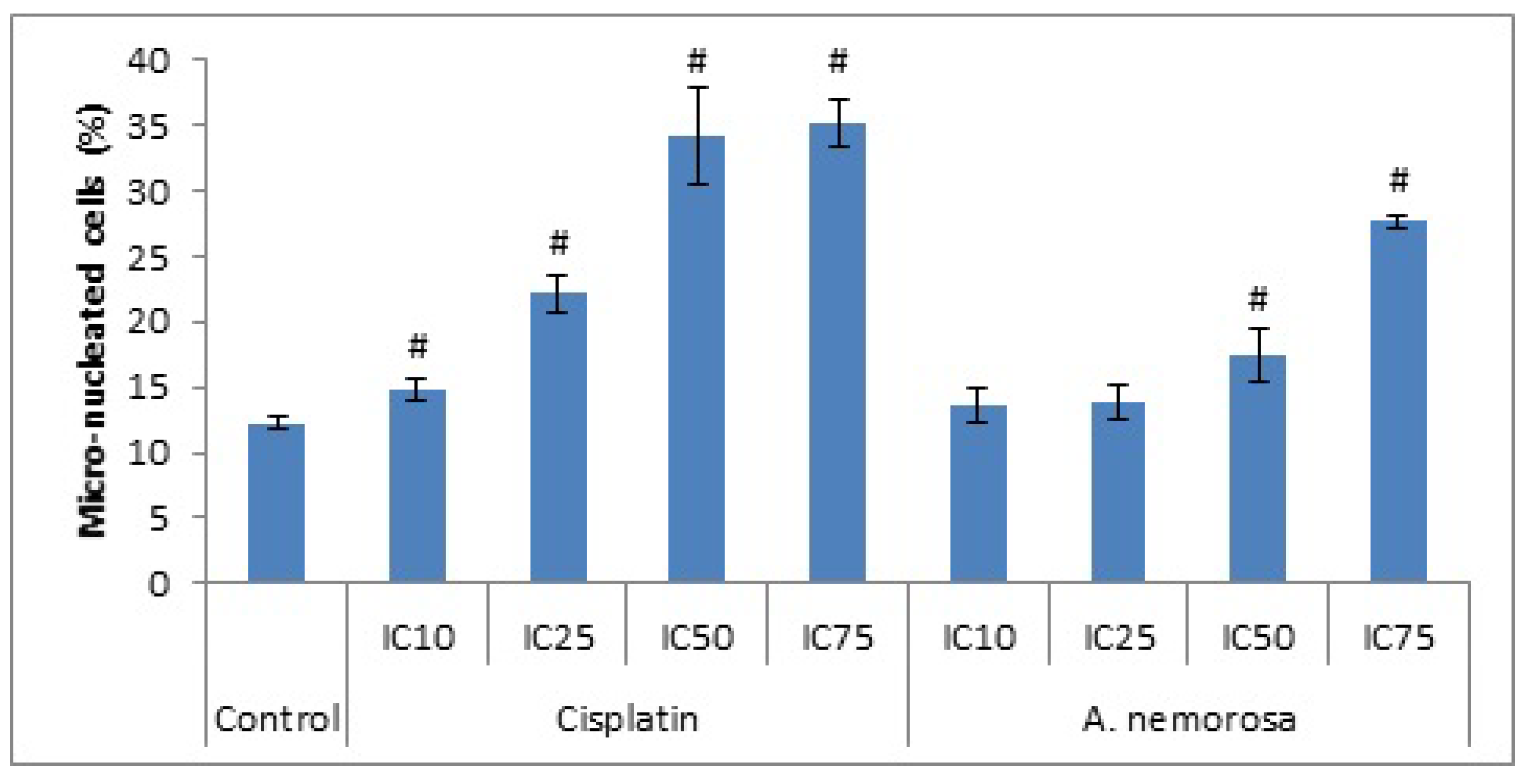
Mitotic catastrophe is a regulated process that uses anti-proliferative measures such as apoptosis, necrosis and senescence during defective or failed mitosis in order to prevent proliferation of cells. Mitotic catastrophe is characterized by the formation of giant multinucleated cells and also micronucleated cells, the former is due to clusters of missegregated uncondensed chromosomes, whereas the latter is as a result of lagging chromosomes or chromosome fragments, during anaphase, which are not incorporated into the daughter nuclei during telophase [
22]. Cell death through mitotic catastrophe can manifest in a caspase-dependent or -independent manner. The caspase-dependent manner involves the release of cytochrome c following mitochondrial membrane depolarization and oligomerization of Bax/Bak. Caspase-independent cell death on the other hand can occur through a sudden Ca
2+ overload, oxidative stress and mitochondrial permeability transition facilitated by the permeability transition pore complex which acts as a bridge between the inner and outer mitochondrial membranes [
9]. After 48 h of treatment with
A. nemorosa significant increases were seen in the formation of micro-nucleated cells which suggests the possible onset of mitotic catastrophe (
Figure 3).
Phosphatidylserine translocation is believed to be an early feature of apoptosis due to the loss of membrane asymmetry upon induction of apoptosis. During apoptosis the membrane phospholipid PS is translocated from the inner to the outer leaflet of the plasma membrane. During late necrosis, the membrane integrity becomes compromised and Annexin V may enter the cell and bind to PS in the inner leaflet. Therefore, staining of cells with Annexin V is commonly used in conjunction with PI. Non-apoptotic or healthy cells would stain negative for both Annexin V and PI whereas cells undergoing early apoptosis would be positive for Annexin V but negative for PI. Late apoptotic/early necrotic cells would stain positive for both Annexin V and PI, whereas necrotic cells would only be positive for PI [
10]. Treatment with
A. nemorosa after 48 h showed a dose dependent increase in apoptotic, late apoptotic/necrotic and necrotic cells. The most significant increases were seen for percentage of apoptotic and late apoptotic/necrotic cells for both the IC
50 and IC
75 treatments (
Figure 5). Cells undergoing necrosis can also stain positive for both Annexin V and PI and therefore it cannot be determined whether or not the cells categorized as late apoptotic/necrotic are actually apoptotic or necrotic, respectively.
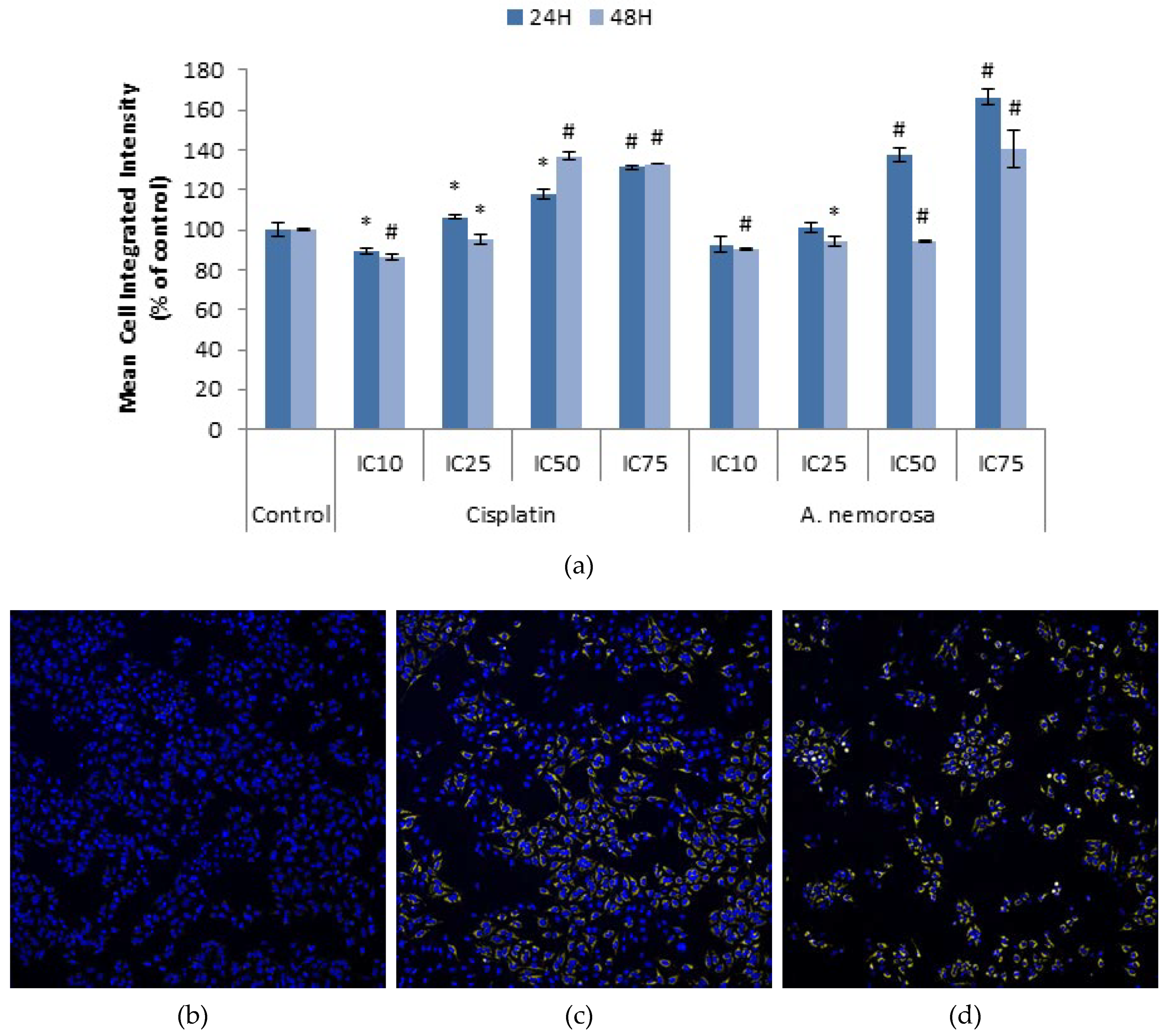
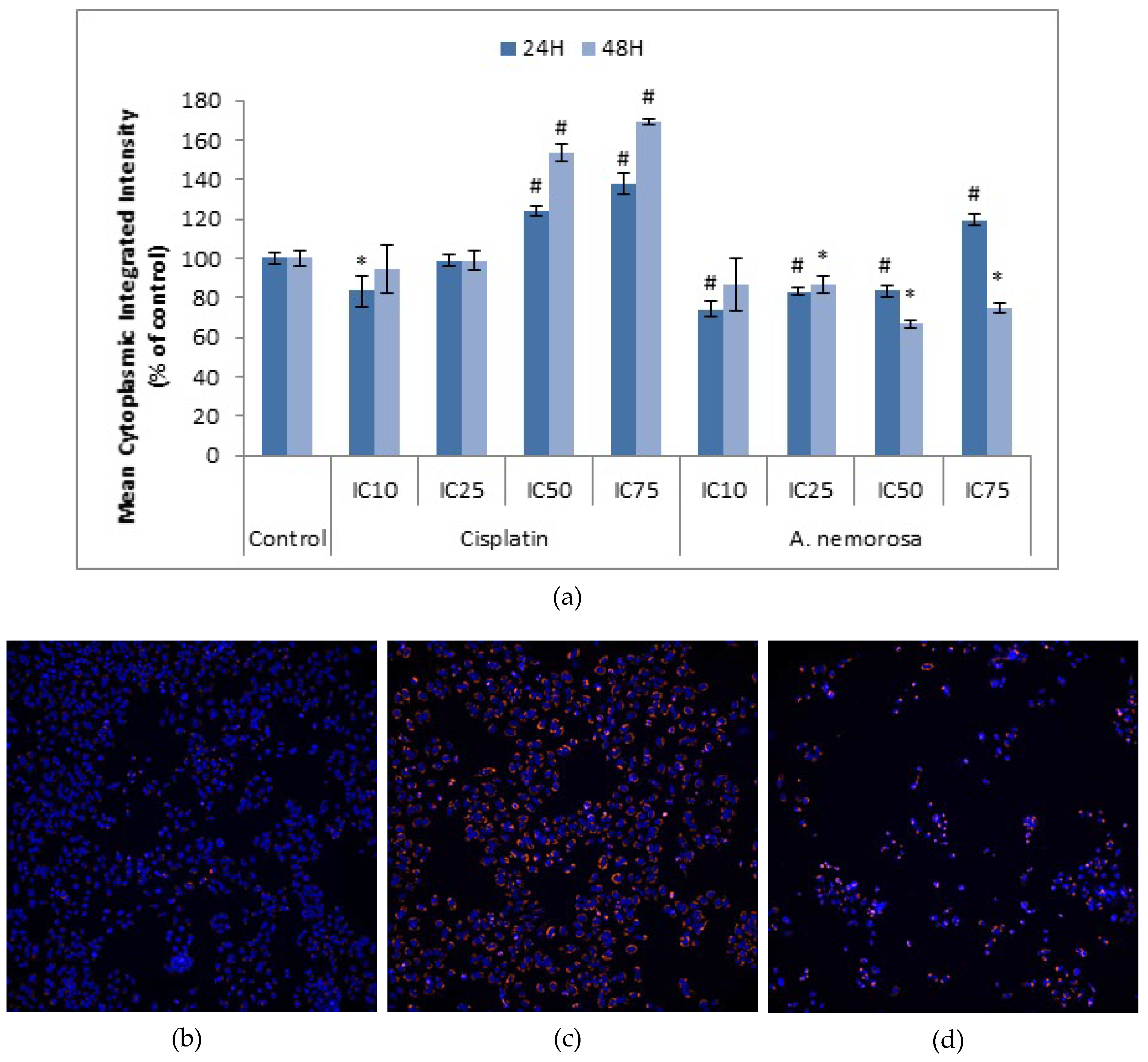
Mitochondrial membrane potential plays a key role in the generation of ATP through the respiratory chain and opening of the mitochondrial permeability transition pore would lead to a collapse of this potential and the subsequent release of cytochrome c (Cyt-
c), Smac/DIABLO, Apoptosis Inducing Factor (AIF) and endonuclease G (Endo G) which all play a role in the initiation of apoptosis either through activation of caspases or leading to DNA fragmentation [
23,
24,
25]. Treatment with
A. nemorosa for 24 and 48 h indicated a decrease in the mean cytoplasmic integrated intensity of TMRE (
Figure 6) and therefore suggests the involvement of the mitochondria in the onset of apoptosis by
A. nemorosa.
Caspases are proteases that contain a cysteine in their active site which cleave in the C-terminal region of aspartate residues and are synthesized as inactive zymogens. Initiator and effector caspases are two groups of caspases involved in cell death. Initiator caspases contain long pro-domains and form aggregates in response to scaffolding co-factors leading to their auto-activation [
26]. In contrast, effector caspases are not capable of auto-activation but become activated through cleavage by initiator caspases. Caspase 8 is an initiator caspase whose activation is promoted by CD95 (Fas/APO-1) and tumor necrosis factor receptor 1 (TNFR1). The active fragments, p18 and p10, of caspase 8 are released during its activation. The activated caspase 8 cleaves and activates effector caspase-1, -3, -6, and -7. Caspase 3 is ultimately responsible for the morphological changes, such as DNA fragmentation and cell shrinkage associated with apoptosis [
27]. The activation of both caspase 8 and caspase 3 without the depolarization of the mitochondrial membrane potential would therefore suggest that the extrinsic pathway plays a role in the execution of apoptosis. Similarly the activation of caspase 3, preceded by mitochondrial membrane depolarization would suggest a role of the intrinsic pathway in the execution of apoptosis [
28]. Significant increases in the mean cell integrated intensities of both caspase 8 and 3 after 24 and 48 h, respectively, was evident in a dose dependent manner (
Table 3 and
Table 4). Thus it can be deduced that the induction of apoptosis by
A. nemorosa involves both the extrinsic and intrinsic pathway. Caspase-dependent apoptosis preceded by mitochondrial membrane depolarization is often associated with increased reactive oxygen species generation. ROS are known to interact with mitochondrial permeability transition complex proteins leading to a significant impact on mitochondrial anion fluxes. The release of cytochrome
c also leads to further ROS increases due to a disrupted electron transport chain [
29]. Treatment with
A. nemorosa significantly increased ROS levels as was evident in the increase in the mean cell integrated intensity (
Figure 6). It can therefore be said that ROS contributes to the induction of apoptosis by
A. nemorosa.
Apoptosis, mitotic catastrophe, autophagy, and regulated necrosis constitute the four major forms of programmed cell death. Cell death is hardly ever due to single pathway activation but rather the result of cross-talk between multiple pathways of which most of the key players participate in more than one signaling cascade. Autophagy is often observed to occur before apoptosis induction, essentially inhibiting it, and is deemed to be a rescue mechanism in which a cell adapts to stress. However, the induction of apoptosis can lead to the cleavage of autophagy-related genes such as ATG3, BECN1, or AMBRA1 which in turn leads to the inhibition of autophagy [
22]. Therefore, the negative or decreased staining observed for treatment with
A. nemorosa (
Figure 7) may be attributed to the absence of autophagy, possibly through inhibition, or it may favor apoptosis induction by tipping the cell survival: cell death balance [
30].
4. Materials and Methods
4.1. Reagents
HeLa cervical cancer cells were purchased from Highveld Biological, Johannesburg, South Africa. RPMI 1640 cell culture medium and fetal bovine serum was purchased from GE Healthcare Life Sciences (Logan, UT, USA). Trypsin-EDTA, Dulbecco’s phosphate buffered saline (DPBS) with Ca2+ and Mg2+ and Dulbecco’s phosphate buffered saline (DPBS) without Ca2+ and Mg2+ were purchased from Lonza (Wakersville, MA, USA). Trypan blue, bisBenzamide H 33,342 trihydrochloride (Hoechst 33342), cisplatin, penicillin/streptomycin and bovine serum albumin fraction V were purchased from Sigma-Aldrich (St. Louis, MI, USA). NucRedTM Live 647, CellRox® Orange reagent, Lysotracker™ Deep Red and Tetramethylrhodamine ethyl ester (TMRE) were purchased from Molecular Probes®-Life Technologies-Thermo Fisher Scientific (Logan, UT, USA). Annexin V-FITC/PI kit was purchased from MACS Miltenyi Biotec (Cologne, Germany). Cleaved caspase 3 (Asp175) (D3E9) Rabbit mAb, Cleaved caspase 8 (Asp391) (18C8) Rabbit mAb, Anti-rabbit IgG (H+L), F(ab’)2 fragment (Alexa fluor® 647 conjugate), Anti-rabbit IgG (H+L), F(ab’)2 fragment (Alexa fluor® 488 conjugate) and Phosphorylated Histone H3 were purchased from Cell Signaling Technology (Danvers, MA, USA).
4.2. Plant Material and Extract Preparation
The aerial part of A. nemorosa was harvested from Piatra Neamţ, Neamţ county (April, 2014) and the identity was established by comparing with herbarium specimens from ‘Dimitrie Brandza’ Botanical Garden, Bucharest. Voucher specimens are available in the herbarium collection of the “Dimitrie Brandza” Botanical Garden, Bucharest (no. 405889) and at the Department of Botany and Cell Biology, “Carol Davila” University of Medicine and Pharmacy, Bucharest.
The aqueous extract of A. nemorosa was prepared by grounding approximately 10 g (mesh 14) and then extracting with 3 × 100 mL water under reflux. This was followed by concentration through rotary evaporation (RVO 004; Ignos, Prague, Czech Republic) and lyophilization at −55 °C (CoolSafe ScanVac 55; LaboGene, Lynge, Denmark), yielding 16.67% dry extract. The reproducibility of the extraction was evaluated on three batches obtained from the same plant material using IR spectra on a JASCO FT/IR-4200 spectrometer with an ATR PRO450-S accessory, on the spectral range of 4000–400 cm−1. For all experiments the plant extract was reconstituted in DMSO at a final concentration of 100 mg/mL and stored at −20 °C until use.
Total polyphenol content (TPC) and total flavonoid content (TFC) were determined according to the Folin-Ciocalteu method at λ = 750 nm and at λ = 429 nm using the aluminum chloride method [
31]. The determinations were performed in triplicate using a UV-VIS spectrophotometer (Halo DB-20-220; Dynamica, Salzburg-Mayrwies, Austria). TPC and TFC were calculated using linear regression by interpolation on calibration curves made in the same conditions and expressed as the means ± SD of the experiments in milligram gallic acid equivalents (GAE) per gram of dry extract (DE) for TPC and in milligram quercetin equivalents (QE) per gram of DE for TFC. TPC was 23.98 mg GAE/g DE, whereas TFC was 1.12 mg QE/g DE.
4.3. Cell Culture Conditions
HeLa cells were routinely maintained in 10 cm culture dishes in RPMI 1640 culture medium supplemented with 10% fetal bovine serum and incubated at 37 °C in a humidified incubator with 5% CO2. Cell number and viability was determined by using a LunaTM cell Counter (Logos Biosystems, Inc., Anyang, South Korea) after staining cells with trypan blue.
4.4. Experimental Imaging and Analysis
For all experiments, unless otherwise indicated, the ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices®, San Jose, CA, USA) was used to image cells. The images were analyzed using the applicable modules of the MetaXpress® High-Content Image Acquisition & Analysis Software supplied by Molecular Devices® (San Jose, CA, USA).
4.5. Cytotoxicity
HeLa cells were seeded in 96-well plates at densities of 5000 cells/well using 100 µL aliquots and left overnight to attach. For treatment, an additional 100 µL of the treatments at varying concentrations of A. nemorosa (0.1–300 µg/mL) and cisplatin (0.1–200 µM) were added. Treated cells were incubated at 37 °C in a humidified 5% CO2 incubator for 48 h. Treatment medium was removed and replaced with 100 µL phosphate buffered saline (PBS with Ca2+ and Mg2+) containing Hoechst 33,342 at a final concentration of 5 µg/mL. PI was added at final a concentration of 10 µg/mL using 10 µL per well of a 110 µg/mL stock and the cells were imaged.
4.6. Cell Cycle Analysis and Micronucleus Assay
HeLa cells were seeded in 96-well plates at a density of 5000 cells/well using 100 µL aliquots and left overnight to attach. The cells were treated with the IC10, IC25, IC50 and IC75 concentrations of A. nemorosa and cisplatin. Treated cells were incubated at 37 °C in a humidified 5% CO2 incubator for 24 and 48 h. Treatment medium was removed and cells were fixed using 4% formaldehyde for 15 min at room temperature prior to the addition of 100 µL NucRedTM Live 647 stain (50 µL NucRed in 10mL PBS, according to manufacturer’s instructions). Cells were incubated for 15 min at room temperature prior to imaging.
4.7. Histone H3 Phosphorylation
HeLa cells were seeded and treated as described for cell cycle analysis. Staining was performed as described for cell cycle analysis, after which cells were permeabilized using 80% ice cold methanol at minus 20 °C for 10 min. Phospho-Histone H3 (Ser10) (D2C8) XP® Rabbit mAb (Cell Signaling Technology, Danvers, MA, USA) was used to determine the presence of phosphorylated Histone H3. After permeabilization the cells were blocked using PBS containing 0.5% BSA and thereafter incubated with the primary antibody (1:250) for 1 h at 37 °C. Cells were washed and incubated with a conjugated secondary antibody (1:1000), anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor® 488 Conjugate) (Cell Signaling Technology, Danvers, Massachusetts, USA) for 30 min at 37 °C in the dark. After staining with the secondary antibody, the cells were washed once to eliminate unbound antibodies and then imaged.
4.8. Phosphatidylserine Translocation
The Annexin V-FITC/PI kit from MACS Miltenyi Biotec (Cologne, Germany) was used with modifications. HeLa cells were seeded and treated as described for cell cycle analysis. After 24 and 48 h, cells were stained by removing treatment medium and adding 50 μL aliquots of a mixture of Annexin V-FITC (50 μL) and Hoechst 33,342 (1 μL) in 5 mL PBS containing 250 μL of Binding Buffer (20×). Cells were incubated in the dark for 15 min at room temperature. PI (50 μL per well of 2 μg/mL stock) was added to the Annexin/Hoechst stain just before image acquisition.
4.9. Mitochondrial Membrane Potential (MMP) Analysis
HeLa cells were seeded and treated as described for cell cycle analysis. Cells were stained by removing treatment medium and adding 100 μL aliquots of a mixture of 0.05 mM tetramethylrhodamine ethyl ester (TMRE) (50 μL) and Hoechst 33,342 (2 μL) in 10 mL PBS. Cells were incubated in the dark for 30 min at 37 °C prior to image acquisition.
4.10. Caspase 8 and 3 Activation
HeLa cells were seeded and treated as described for cell cycle analysis analysis. Cells were treated for 24 and 48 h, after which they were fixed and permeabilized as described for histone H3 phosphorylation. Cleaved caspase 8 (Asp391) and cleaved caspase 3 (Asp175) (D3E9) rabbit monoclonal antibodies (Cell Signaling Technology) were used to determine the presence of activated caspase 8 and caspase 3, respectively. After permeabilization the cells were blocked using PBS containing 0.5% BSA and thereafter incubated with the antibodies separately (1:100 for caspase 8 and 1:200 for caspase 3) for 1 h at 37 °C. Cells were washed and incubated with a conjugated secondary antibody (1:500), anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor® 647 Conjugate) for 30 min at 37 °C in the dark. After staining with the secondary antibody, the cells were washed once to eliminate unbound antibodies and Hoechst 33,342 was used as a counterstain prior to imaging.
4.11. Reactive Oxygen Species (ROS) Production
HeLa cells were seeded and treated as described for cell cycle analysis. Cells were stained by removing treatment medium and adding 100 μL aliquots of a mixture of 2.5 mM CellRox orange (20 μL) and Hoechst 33,342 (2 μL) in 10 mL PBS. Cells were incubated in the dark for 30 min at 37 °C prior to image acquisition.
4.12. Autophagy Induction
HeLa cells were seeded and treated as described for cell cycle analysis. Cells were stained by removing treatment medium and adding 100 μL aliquots of a mixture of 50 nM Lysotracker red (0.5 μL) and Hoechst 33,342 (2 μL) in 10 mL PBS. Cells were incubated in the dark for 30 min at 37 °C prior to imaging.
4.13. Statistical Analysis
At least three experiments were completed for each cell line in which three different transfer numbers were used. Statistical analysis was performed by means of the student t-test for two samples assuming equal variance. Error bars represent the standard deviation of the mean (SD).


 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}