1. Introduction
Sepsis is estimated to be one of the leading causes of critical illness and mortality with constantly increasing incidence in intensive care units [
1,
2]. Sepsis is defined as an organ dysfunction of variable severities induced by a dysregulated immune response to an infection [
3,
4]. One of the organs determining early and late outcomes of sepsis is the kidney. Septic acute kidney injury (AKI) can be established within hours to days in up to 50% of intensive care unit patients [
5]. In surviving patients, even mild or short episodes of AKI can predispose to an increased risk for developing chronic kidney disease and end-stage renal failure [
4].
The acute phase response/reaction (APR) is part of the immune response to infections and tissue damage. Proteins whose plasma concentration is changed by at least 25% in response to pro-inflammatory stimuli are termed acute phase proteins (APP) [
6,
7,
8]. They have a role in restoring homeostasis after inflammation [
7]. Although APPs are thought to be primarily produced by the liver and secreted into the blood, they are also synthesized in other organs. Thus, APPs can contribute to local defense responses and repair mechanisms [
8].
Plasma concentrations of APPs (e.g., C-reactive protein) are well-established markers for diagnosing and monitoring sepsis [
9]. Lipocalin-2 (Lcn-2) also termed as neutrophil gelatinase-associated lipocalin (NGAL) is a sensitive marker of renal tubular injury [
10,
11,
12,
13]. However, none of these biomarkers is specific for sepsis or AKI [
9,
14]. For example, elevated serum Lcn-2 concentrations were observed in other pathologic conditions, such as cardiovascular [
15] and gastrointestinal [
16,
17] diseases. Several other AKI biomarker candidates have been identified [
14,
18] but so far none of them was able to differentiate the various types of AKI with satisfactory specificity and sensitivity [
14,
19]. For this reason, there is an intensive effort to identify biomarkers or biomarker combinations that could be used for the early detection or follow-up of AKI [
14].
The inflammatory and circulatory effects of sepsis are often modelled in rodents by administration of bacterial wall endotoxin (lipopolysaccharide: LPS) [
20,
21]. Increased renal production of some APPs, such as fibrinogen [
22], ceruloplasmin [
23], complement C3 [
24,
25], haptoglobin [
26,
27,
28], hemopexin [
29], serum amyloid A [
23,
27], beta-2-microglobulin [
22], α-1-acid glycoprotein [
23], and plasminogen activator inhibitor-1 [
30] have been described in this model, while renal gene expression of serum albumin (a negative APP) was shown to be reduced [
31]. Although these scarce and isolated findings demonstrate the renal production of some APPs in sepsis, a coordinated, complex APR of the kidney has not been evaluated before.
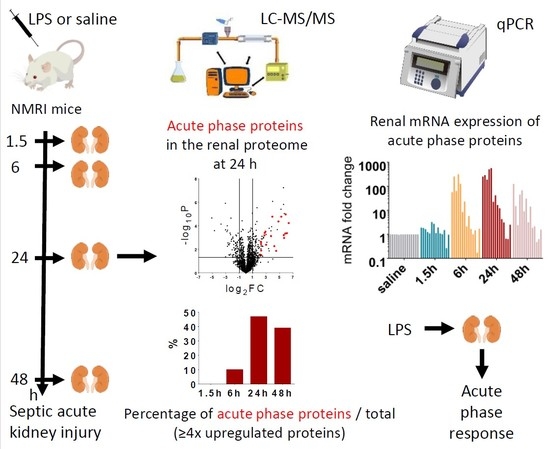
In the present study, we performed proteomic analysis to find relevant groups of proteins activated by LPS at early (1.5 h, 6 h) and late (24 h, and 48 h) time points after endotoxin administration in the kidney. Surprisingly, we found abundant representation of APPs among the most regulated proteins. Furthermore, qPCR measurements revealed that 14 out of these APPs were produced locally, in the injured kidney.
2. Results
2.1. LPS-Induced Severe Inflammation in the Kidney
All animals survived in the EP groups (40 mg/kg LPS), but one mouse died both in the LP24 and LP48 groups (10 mg/kg LPS) during the experiment.
Endotoxin significantly upregulated renal TNF-α and IL-6 mRNA production at all time points (
Figure 1). Both LPS doses caused severe, maximal inflammatory response in the kidneys. IL-6 mRNA but not TNF-α mRNA started to decrease at 48 h.
2.2. LPS-Induced Tubular Damage in the Kidney
LPS significantly upregulated renal Lcn-2 mRNA and protein expression already from 1.5 h and from 6 h, respectively (
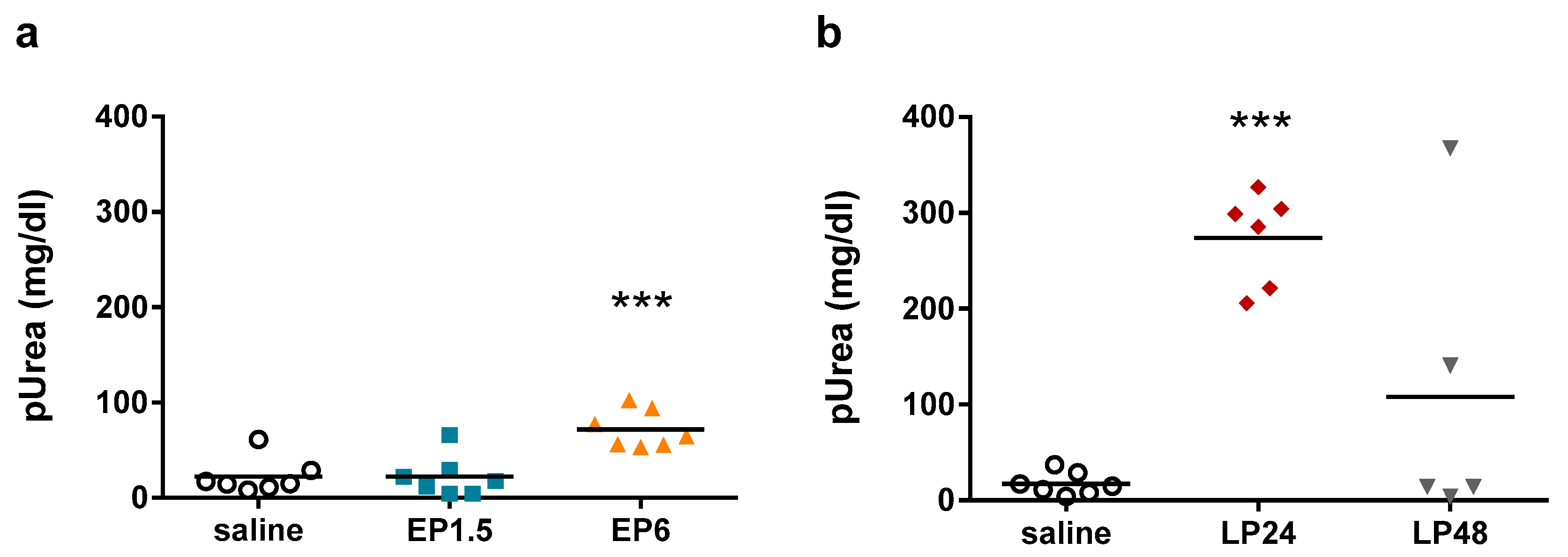
Figure 2), indicating tubular injury. Plasma urea concentrations were elevated first at 6 h after LPS administration, increased further at 24 h despite the lower LPS dose and started to decrease at 48 h (
Figure 3), indicating impaired renal function.
The decrease in both Lcn-2 mRNA and plasma urea concentrations at 48 h indicate reversibility of septic AKI in our experimental setting.
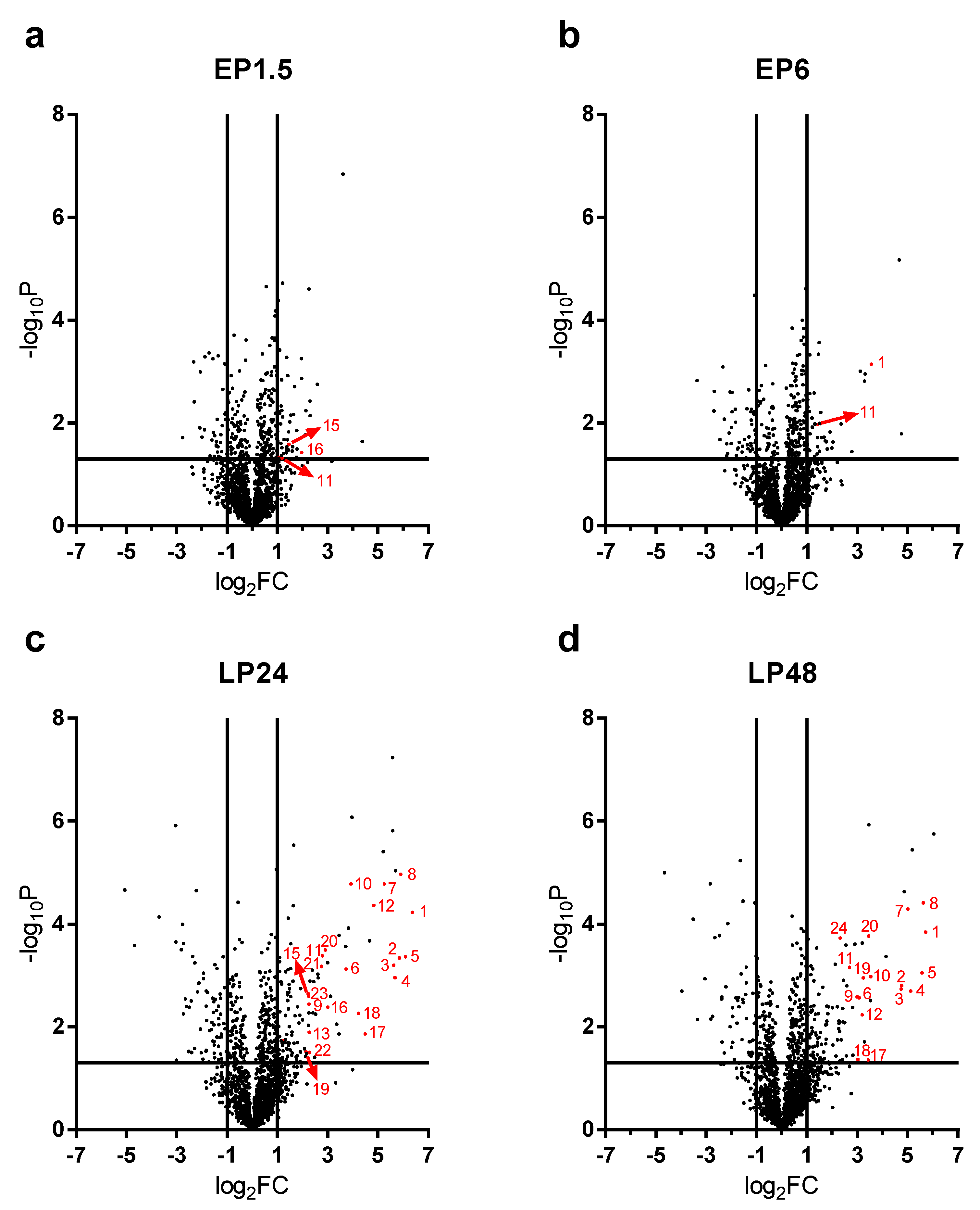
2.3. Renal Protein Concentration Changes at Early Phases of AKI
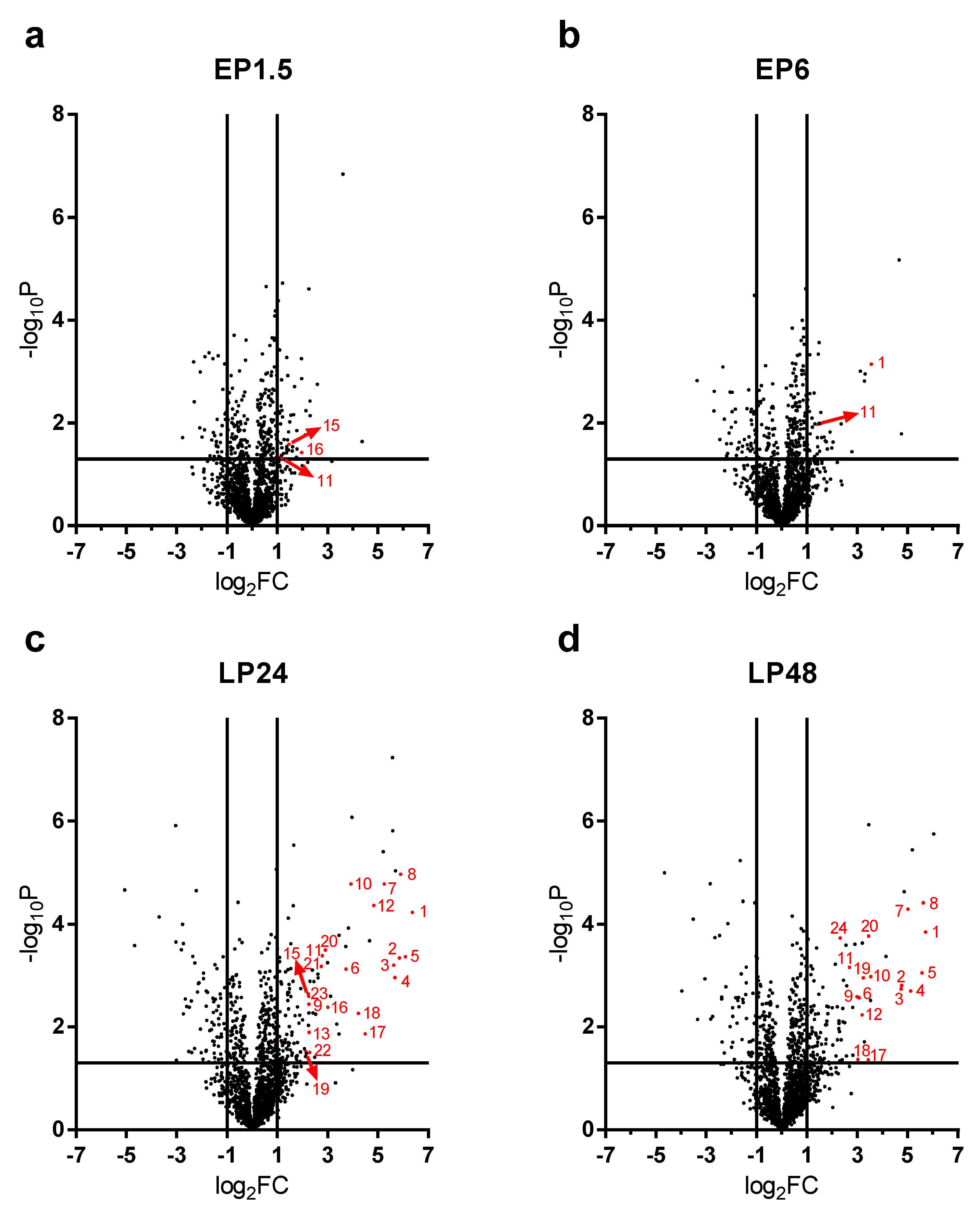
LPS-induced renal proteome effects became highly significant and abundant, involving many proteins first at 24 h after LPS injection (
Figure 4c). Only 10-10 proteins were upregulated at least 4-fold (log
2FC = 2) in the EP1.5 and EP6 groups (
Figure 4a,b) and the changes were smaller in the EP than LP groups. Several proteins were upregulated at both time points (
Table 1, grey highlights) with quite similar fold changes and rankings. In EP, only Lcn-2 was upregulated more than 4-fold out of the APPs.
2.4. Acute Phase Proteins Were the Most Upregulated Proteins in the Kidney in the Late Phase After LPS Administration
As opposed to the moderate proteome changes observed in EP, forty-seven and forty-four proteins were upregulated at least 4× (log
2FC = 2) at LP24 and LP48, respectively. The first 20 hits are shown in
Table 2, the remaining 27 and 24 hits are shown in
Supplementary Table S1. Just as in EP, about 2/3 of the proteins were upregulated at both time points, and several proteins were upregulated in both LP and EP.
APPs were abundantly present among the significantly upregulated proteins in LP (
Figure 4c,d). At LP24 forty-seven %, while at LP48 thirty-nine % of the identified proteins were APPs. Of the 20 top hits, 12-12 proteins were APPs at LP24 and LP48 (
Table 2, bold highlight).
As the top 20 hits included APPs in such a high proportion at LP we reanalyzed the MS data of APPs and collected all the significantly upregulated APPs in
Table 3. The list is led by the 3 chains of fibrinogen and complement C3, followed by transport proteins (e.g., ceruloplasmin, haptoglobin, hemopexin, transferrin, ferritin heavy chain) and amyloids, protease inhibitors (inter alpha-trypsin inhibitors, alpha-2-macroglobulin) and serine protease inhibitors (serpins).
Lipocalin-2 (an established AKI marker) appeared third, first and second on the list of the upregulated proteins at EP6, LP24 and LP48, respectively.
The following APPs were enriched in the kidneys after LPS administration in both LP groups: complement C3, fibrinogen-α, -β, -γ, two isoforms of serum amyloid A (Saa1 and Saa2), ceruloplasmin, haptoglobin, hemopexin, inter alpha-trypsin inhibitor heavy chain 4, transferrin, serine protease inhibitor A3K (Serpina3k), alpha-2-macroglobulin (A2m) and beta-2-microglobulin (B2m). Apolipoproteins A1 (ApoA1) and E (ApoE), inter-alpha-trypsin inhibitor heavy chain H1 (Itih1), vitamin D-binding protein or Gc-globulin (DBP) and serine protease inhibitor A3N (Serpina3n) were significantly elevated only in LP24, while alpha-1-acid glycoprotein (A1AGP) and von Willebrand factor A domain-containing protein 5A (Vwa5a) were upregulated only in LP48.
Renal ferritin heavy chain protein was significantly elevated at all 4 time points. Alpha-1-antitrypsin (Serpina1) protein was significantly elevated in EP1.5, LP24 and LP48. Albumin protein fluctuated, it was significantly increased in the EP1.5 and LP24 groups, while it was at control level in EP6 and LP48.
2.5. Most of the Upregulated Proteins Were Related to Inflammation in the Late Phase
In addition to the APPs, most of the other proteins upregulated at least 4× in LP were also involved in stress responses based on Gene Ontology (GO) analysis (
Table 4). At LP24 sixty % and at LP48 fifty-six % of the top ranking proteins belonged to the ‘response to stress’ category. Among the top 20 hits (
Table 2) almost all proteins were either APPs or proteins involved in stress response (highlighted in
italics). Definitions of the biological process categories are on the GO website (
http://www.informatics.jax.org/vocab/gene_ontology/).
2.6. Acute Phase Protein Synthesis Was Stimulated in the Kidney after LPS
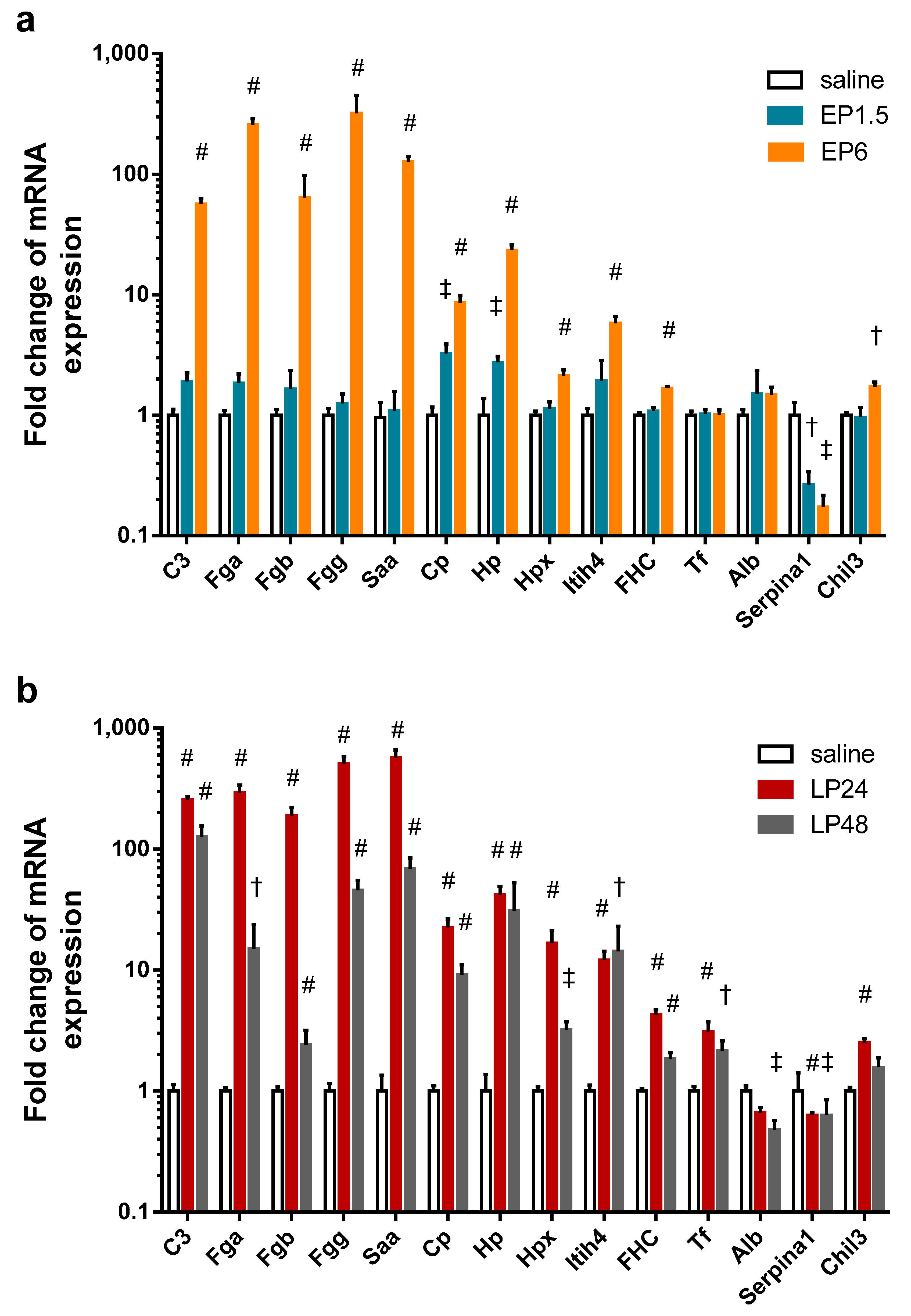
To verify the source of the APPs found in the kidney with MS, we conducted quantitative PCR analysis and detected that LPS significantly upregulated the renal mRNA expression of several APPs at EP6 and LP (
Figure 5): complement C3, fibrinogen-α, -β, -γ, serum amyloid A, ceruloplasmin, haptoglobin, hemopexin, inter alpha-trypsin inhibitor heavy chain 4, and ferritin heavy chain. Only ceruloplasmin and haptoglobin mRNA were elevated already at 1.5 h. Transferrin expression was upregulated in the kidneys only in LP.
Fibrinogen-α, -β, -γ, serum amyloid A, ceruloplasmin, hemopexin and ferritin heavy chain mRNA decreased at 48 h. Although complement C3 and transferrin mRNA also showed a decreasing tendency in the LP48 group, the difference was not statistically significant.
Albumin expression was unchanged in the kidneys during EP, but showed a decreasing tendency in LP24 with significant downregulation at 48 h.
As the MS analysis did not differentiate between two isoforms of alpha-1-antitrypsin (Serpina1a and Serpina1c), we measured the mRNA expression of these two isoforms separately with primers amplifying only one of them. Neither isoform was expressed in any of the kidney samples. As Zager et al. [
32] could amplify alpha-1-antitrypsin in renal tissues, using their primers we were also able to amplify alpha-1-antitrypsin mRNA, however, it was downregulated in all LPS-administered groups. As this primer also amplifies the 1d isoform in addition to 1a and 1c isoforms, the decreased mRNA expression could be attributed to the Serpina1d isoform, not detected by the MS.
Serine protease inhibitor A3K mRNA was undetectable in all kidney samples.
Another inflammatory marker, chitinase-like protein 3 mRNA was upregulated in the EP6 and LP24 groups and still upregulated, but to a less extent, at 48 h.
3. Discussion
APP production is a well-known consequence of inflammatory stimuli such as sepsis or LPS. However, the general view is that the main source of plasma APPs is the liver [
8,
33]. Although production of some APPs in the kidney have been demonstrated before, this is the first comprehensive demonstration of a massive, combined renal APR in sepsis.
The LPS-induced AKI was verified by upregulated TNF-α, IL-6, and Lcn-2 mRNA expression in the kidneys. IL-6 and Lcn-2 mRNA started to decline together with several other factors at 48 h after endotoxin administration, indicating the start of recovery.
The fact that several proteins were included on at least 2 lists (both EP and LP) with quite similar fold changes and rankings supports the reliability of our MS analysis.
Lipocalin-2 (also called as neutrophil gelatinase associated lipocalin: NGAL) is a well-known marker of AKI [
34]. It appeared among the upregulated proteins first on the 6-h list and ranked first on the LP24 and second on the LP48 lists, supporting the diagnostic value of Lcn-2 as an AKI marker.
The majority of the mostly upregulated proteins was related to stress and inflammation. For example, the interferon response is a well-known mechanism activated by viral and bacterial infections [
35,
36]. Correspondingly, interferon-inducible proteins appeared together with APPs in the LP lists.
Regarding the renal APR, our results are also in line with scarce previous findings demonstrating that endotoxin upregulated renal mRNA and/or protein expression of haptoglobin [
26,
27,
28], hemopexin [
29], ceruloplasmin [
23], serum amyloid A [
23,
27], complement C3 [
24,
25], and all three chains of fibrinogen [
22] 24 h after LPS administration, while albumin was downregulated [
31]. We identified additional APPs upregulated by LPS in the kidney such as inter alpha-trypsin inhibitor heavy chain 4, transferrin, ferritin heavy chain, alpha-2-macroglobulin, apolipoprotein A1, apolipoprotein E, vitamin D-binding protein, inter-alpha-trypsin inhibitor heavy chain H1, serine protease inhibitor A3K, A3N, and alpha-1-antitrypsin. Although transferrin is a negative APP in humans, it is regarded as a positive APP in mice [
37].
The identified highly upregulated APPs (coagulation system components, complement C3, transport proteins, amyloids and protease inhibitors) have been considered to be primarily produced by the liver [
33]. However, several APPs have been demonstrated to be produced in other organs under physiological conditions. Examples include haptoglobin expression in the skin and lung [
8], transferrin in the brain [
38], ceruloplasmin in the kidney, mammary gland and brain [
39], hemopexin in the nervous system and retina [
40], complement C3 in the immune cells and the kidney [
41], and serum amyloid A in many tissues including the kidney [
42]. Ferritin is expressed in all organs including the kidneys [
43]. In general, APPs are expressed at a low level in extrahepatic tissues under physiologic conditions but can be induced by local injury [
8].
Chitinase-like protein 3 (Chil3) was also upregulated in the kidney. It is a rodent-specific member of the chitinase-like protein family. It is a chemoattractant for eosinophils and a marker of murine M2 macrophages and N2 neutrophils [
44,
45,
46,
47]. Another, highly homologous member of the chitinase-like protein family, chitinase-3-like protein 1 (CHI3L1) is an APP in humans [
48]. Both CHI3L1 and Chil3 were elevated in the serum of septic mice [
49], suggesting that Chil3 may also be involved in the APR.
The role of this endotoxin-associated renal APR is probably homeostatic, by providing tissue protection [
7]. Some APPs are effectors of the immune system (e.g., complement C3 [
50]), some recruit immune cells (e.g., fibrinogen [
51], serum amyloid A [
52] and chitinase-like protein 3 [
44]), some have antioxidant or anti-inflammatory functions (e.g., ceruloplasmin [
53], hemopexin [
54], haptoglobin [
55], inter alpha-trypsin inhibitor heavy chain 4 [
56], alpha-1-acid glycoprotein [
57], and apolipoproteins [
58,
59]), some protect tissues from proteolytic destruction (e.g., alpha-1 antitrypsin [
60] and alpha-2-macroglobulin [
61]), some have cytoprotective scavenger functions (e.g., ferritin heavy chain [
62] and transferrin [
63] bind free Fe, haptoglobin sequesters hemoglobin [
55], hemopexin binds heme and bivalent metal ions [
54], apolipoprotein A1 [
58] and vitamin D-binding protein [
64] neutralize LPS), and some initiate tissue repair (e.g., fibrinogen [
50]).
A likely role of the APPs produced in the kidney is mediating cell survival [
65]. For example, the heme-hemopexin complex [
54] and ferritin heavy chain [
66] have been shown to inhibit apoptosis. The role of ferritin heavy chain has already been studied in different AKI settings. Rhabdomyolysis, cisplatin-induced AKI and unilateral ureteral obstruction all had worse outcomes in proximal tubule-specific ferritin heavy chain knockout mice [
67,
68]. Furthermore, overexpression of ferritin heavy chain in the kidneys prior to ischemia-reperfusion injury had cytoprotective effects [
69]. It has also been demonstrated that sublethal doses of LPS can protect the kidneys from a subsequent lethal damage such as severe ischemia-reperfusion injury [
70,
71]. If APPs indeed provide protection to the kidney, they may be functionally involved in the mechanism of LPS-induced preconditioning.
Although most APPs serve to preserve tissue integrity, some of them can mediate both protection and injury. For example, fibrinogen can enhance inflammation by activating immune cells, while it initiates tissue repair as a scaffold for tissue reconstruction [
51,
72,
73]. In fibrinogen knockout mice, renal fibrogenesis was reduced after unilateral ureteral obstruction [
74], but renal function and survival was worse following ischemia-reperfusion injury [
75].
Serum amyloid A also mediates both proinflammatory and anti-inflammatory processes. It can attract phagocytes and enhance proinflammatory cytokine production but can promote anti-inflammatory cytokine production and polarisation of macrophages to M2 phenotype [
52,
76]. Similarly, ceruloplasmin is also known to have both prooxidant and antioxidant activities [
53].
The markedly enhanced renal production of a great number of APPs puts them forward as biomarker candidates. Zager et al. [
28] found that LPS induced greater upregulation of haptoglobin in the kidneys than in the liver in mice, and they detected elevated urinary haptoglobin in patients with AKI. Also, chitinase-like proteins, including Chil3 were suggested to be potential biomarkers of septic AKI in both mice and human [
49].
However, before any APP is considered as a biomarker of septic AKI, their timely expression pattern should be determined. Most previous studies determined changes in APPs solely 24 h after endotoxin administration. Only scarce previous findings are available regarding EP, e.g., haptoglobin was upregulated [
28], while albumin was unchanged [
31], and hemopexin was also slightly increased 4 h after LPS administration in mice [
29].
Taken together, renal gene expression of many APPs was induced already during the EP and peaked at 24 h. Among the APPs studied, ceruloplasmin, haptoglobin, and Lcn-2 mRNA were upregulated earliest. They were followed at 6 h by complement C3, fibrinogen-α, -β, -γ, hemopexin, serum amyloid A, inter alpha-trypsin inhibitor heavy chain 4, and ferritin heavy chain. All these gene expression changes in EP translated into elevated protein levels in the kidneys at the established phase at 24 h. Only ferritin heavy chain protein was already elevated in EP. Transferrin responses were delayed as both mRNA and protein appeared elevated first at LP24. Reduction of the mRNA expression of fibrinogen-α, -β, -γ, serum amyloid A, ceruloplasmin, hemopexin, and ferritin heavy chain by 48 h vs. 24 h, suggests initiation of recovery.
As a negative APP, renal albumin mRNA was suppressed by LPS during LP. The increase in renal serine protease inhibitor A3K and alpha-1-antitrypsin isoforms Serpina1a and 1c were not the result of renal expression. We hypothesize that their source may be the liver, since serine protease inhibitor A3K is a liver-specific gene [
77]. Based on our data, none of the kidney specific APPs are earlier markers of septic AKI than Lcn-2.
4. Materials and Methods
4.1. Animal Studies
Male Naval Medical Research Institute (NMRI, Bethesda, MD, USA) mice (Hsd:Win:NMRI mice (RRID_MGI_MGI:6198565); Toxi-Coop Ltd., Budapest, Hungary) weighing 25–30 g were housed under standard conditions with free access to food and tap water. All protocols were approved by the Pest County Government Office and the Animal Ethics Committee of Semmelweis University (code: PE/EA/2202-5, date: 14 December 2017).
4.2. LPS Application
Mice were injected with LPS (0111:B4; Sigma-Aldrich, Budapest, Hungary) at the doses of 10 or 40 mg/kg bodyweight (BW) intraperitoneally (i.p.). These maximal doses were selected based on our previous experiments to ensure septic shock but no lethality throughout the follow-up period [
70,
71]. Two time points represented the early phase (EP, 1.5 and 6 h), while the other two represented the late phase (LP) of LPS-induced inflammation and AKI (24 and 48 h) [
78]. LPS was freshly suspended in sterile saline immediately before use. Animals were divided into four treatment groups as follows:
Group 1 (EP1.5): LPS at 40 mg/kg BW, and sacrificed at 1.5 h post-injection (n = 7)
Group 2 (EP6): LPS at 40 mg/kg BW, and sacrificed at 6 h post-injection (n = 7)
Group 3 (LP24): LPS at 10 mg/kg BW, and sacrificed at 24 h post-injection (n = 7)
Group 4 (LP48): LPS at 10 mg/kg BW, and sacrificed at 48 h post-injection (n = 7)
Mice (n = 7/each dose) received equal volumes of saline to serve as controls.
4.3. Organ Harvest
Mice were injected with 5000 U/kg BW heparin i.p. (Ratiopharm GmbH, Ulm, Germany) and 3 min later they were sacrificed by cervical dislocation. The chest was opened and blood was collected from the thoracic cavity after cross-section of the vena cava. Blood was removed from parenchymal organs and the whole circulation by injecting 10 mL 4 °C saline transcardially. The kidneys were removed, decapsulated and cut into pieces. One piece from each kidney was placed in 500 µL TRI Reagent (TR 118, Molecular Research Center, Inc., Cincinnati, OH, USA). Together with the parts of the kidney were snap frozen in liquid nitrogen and kept at −80 °C for RNA isolation and proteomic analysis.
Livers were also collected similarly, from intact NMRI mice for control purposes.
4.4. Plasma Urea Determination
Plasma urea concentrations were measured by a urease and glutamate-dehydrogenase enzymatic assay with colorimetric detection according to the manufacturer’s protocol (Diagnosticum Zrt., Budapest, Hungary). The urea concentration of the samples was determined using a standard curve.
4.5. Tissue Homogenization
Four kidney samples selected as most representative from each treatment group and four control kidney samples were processed for mass spectrometry analysis as described before [
79] with a slightly modified protocol. Frozen tissues were cut into pieces of about 30 mg on dry ice to prevent thawing. Next, 8 µL lysis buffer/mg of tissue was added to each piece. The lysis buffer contained 50 mM Tris pH 8.0 (SERVA Electrophoresis GmbH, Heidelberg, Germany), 150 mM NaCl (Thermo Fisher Scientific, San Jose, CA, USA), 1 mM EDTA (SERVA Electrophoresis GmbH, Heidelberg, Germany), 1% NP-40 lysis buffer (Roche, Basel, Switzerland), 0.5% Na-deoxycholate (Sigma-Aldrich, Maribor, Slovenia), 0.1% sodium dodecyl sulfate (Pierce Chemical, Dallas, TX, USA) and 1% protease inhibitor cocktail (Sigma-Aldrich, Maribor, Slovenia). The tissue was homogenized by Dounce homogenizer, and then repeatedly passed through a 0.7 × 38 mm syringe needle for 20 times for further homogenization. The samples were then gently shaken for 30 min on ice (Rotamax 120, Heidolph Instruments GmbH, Schwabach, Germany). After centrifugation (16,000
g, 10 min), the protein concentration in the supernatant was determined by the Bradford assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and 150 µg of total protein from each sample was incubated with loading buffer (10 mM Tris pH 6.8; SERVA Electrophoresis GmbH, Heidelberg, Germany); 7.8% glycerol (CARLO ERBA Reagents, Val de Reuil, France); 0.2% SDS (Pierce Chemical, Dallas, TX, USA); 0.01% Bromphenol blue (Sigma-Aldrich, Maribor, Slovenia)) containing 10 mM dithiothreitol (DTT) (Fluka Biochemica, Steinheim, Germany) at 95 °C for 5 min. The samples were stored at −20 °C until further processing.
4.6. Sample Preparation for Mass Spectrometry
Sample preparation for LC-MS/MS analysis was performed as described previously [
80]. Briefly, 150 µg of total protein from each sample was separated on a 12.5% SDS-PAGE gel (Lonza, Basel, Switzerland). The gel was stained with Coomassie brilliant blue and each of the protein lanes was cut into six bands and destained with 25 mM ammonium bicarbonate in 50% acetonitrile. The gel pieces were washed with acetonitrile, vacuum dried and rehydrated in reducing solution (10 mM DTT; Fluka Biochemica, Steinheim, Germany), 25 mM NH
4HCO
3. After 45-min incubation at 56 °C, solution was exchanged to alkylating solution (55 mM iodoacetamide; Amersham Biosciences, Little Chalfont, UK), 25 mM NH
4HCO
3 and samples were incubated in the dark at room temperature for 30 min. The gel pieces were washed with acetonitrile and vacuum dried before rehydrating in 80 µL of trypsinization buffer (25 mM NH
4HCO
3) containing 1 µg of sequencing-grade modified porcine trypsine (Promega, Madison, WI, USA) per sample. After overnight incubation at 37 °C, the trypsin solution was collected and remaining peptides were extracted from the gel pieces using extraction solution (50% acetonitrile, 5% formic acid; JT Baker, Center Valley, PA, USA). Trypsin solution was added to extraction solution and concentrated by vacuum drying to a final volume of about 20 µL.
4.7. Mass Spectrometry Analysis
LC-MS/MS analysis was performed with an EASY-nanoLC II HPLC unit (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Orbitrap LTQ Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Samples containing 0.1% FA were loaded onto a C18 trapping column (Proxeon Easy-column, Thermo Fischer Scientific, West Palm Beach, FL, USA) and separated on a C18 PicoFrit Aquasil analytical column (New Objective, Inc., Woburn, MA, USA). The peptides were eluted using a 5–40% (v/v) 90 min linear gradient of acetonitrile in a 0.1% formic acid solution at a constant flow rate of 300 nL/min. The full MS mass spectra were acquired with the Orbitrap mass analyzer in the mass range of 300 to 2000 m/z at resolution of 30,000. The MS/MS spectra were obtained by HCD fragmentation of the nine most intense MS precursor ions and recorded at a resolution of 7500. Only the precursor ions with assigned charge states (> 1) were selected for MS/MS fragmentation. The dynamic exclusion was set to repeat count of 1, repeat duration of 30 s, and exclusion duration of 20 s.
4.8. Data Analysis
Data analysis was performed as described previously [
80] with minor modifications. MaxQuant proteomics software (version 1.6.0.13; Max-Planck Institute for Biochemistry, Martinsried, Germany) was used for database search and quantification by spectral counting [
81]. Database search was performed against a Mus musculus Uniprot database (database date 15.10.2017, 16,923 entries). For the database searches methionine oxidation (+15.995 Da) and protein N-terminal acetylation (+45.011 Da) were set as variable modifications. Carbamidomethylation of cysteines (+57.021 Da) was set as fixed modification. Trypsin cleavage after arginine and lysine was used as the enzyme specificity. For database searches one missed cleavage was allowed. In addition, precursor ion and fragment ion mass tolerances were set to 20 ppm and 0.5 Da, respectively. A reversed database search was performed and the false discovery rate was set at 1% for peptide and protein identifications. Raw data and database search files are available via ProteomeXchange with identifier PXD014664 [
82]. Relative quantification of identified proteins was performed by label-free quantification (LFQ) algorithm in MaxQuant.
4.9. RNA Preparation
Total RNA was extracted from the kidneys and livers with TRI Reagent (TR 118, Molecular Research Center, Inc., Cincinnati, OH, USA) according to the protocol provided by the manufacturer. RNA concentration and purity was checked with NanoDrop 2000 c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and RNA integrity was verified by electrophoresing the samples on 1% agarose gel (Invitrogen Ltd., Paisley, UK). The RNA solutions were stored at −80 °C until further procedures.
4.10. qPCR Analysis of Gene Expression
The gene expression of MS identified APPs assessed by qPCR are summarized in
Table 5. Each primer was tested on normal liver samples as positive control. TNF-α and IL-6 mRNA levels were determined to assess inflammation induced by LPS administration in the kidneys of mice. Renal tubular damage was assessed based on Lcn-2 gene expression. The endogenous reference gene was GAPDH.
Messenger RNA (mRNA) levels were measured as described previously [
10]. Reverse transcription into cDNA was carried out by the High-Capacity cDNA Archive Kit (Applied Biosystems™, Foster City, CA, USA) according to the manufacturer’s protocol. In brief, 1 μg of total RNA was denaturized at 70 °C for 5 min. After the annealing of the random hexamer primers on the RNA template at 25 °C for 10 min, cDNA was synthesized at 37 °C for 2 h. The reaction was terminated by heat inactivation (85 °C for 2 min). Gene expression from kidney tissue homogenates was evaluated on the Bio-Rad C1000™ Thermal Cycler with CFX96™ Optics Module real-time PCR system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The PCR reaction was performed with SensiFAST™ SYBR No-ROX Kit (Bioline Reagents Limited, London, UK), according to the manufacturer’s protocol. Primers (
Table 5) were designed by NCBI/Primer-BLAST online software with the exception of the Serpina1 primer that was adapted from Zager et al. [
32] Primers were synthesized by Integrated DNA Technologies. Primer annealing was set to 60 °C. All samples were measured in duplicates and expression was calculated using the relative quantification (ΔΔ
Cq) method. The efficiency of the qPCR reaction was verified with standard curves. The melting curve was also viewed to detect any abnormality of the PCR product. Expression was considered to be below the limit of detection in the samples in which no specific amplification product was present.
One mouse in the LP48 group was excluded from the statistical analysis because most of its mRNA expression values were identified as outliers within the group (ROUT method).
4.11. Protein Classification
Gene Ontology Enrichment Analysis powered by PANTHER [
83,
84,
85,
86] was performed to assign proteins to gene ontology biological function categories. Proteins were classified as APPs based on the comprehensive list of classical and putative APPs by Salgado et al. [
87].
4.12. Statistical Analysis
All continuous data are expressed as mean ± standard error of the mean (SEM), unless otherwise stated. LFQ intensity values were log2 transformed for statistical analysis. Fold changes (FC) were determined as the difference of the means between the treated and control groups (log
2FC). Messenger RNA fold changes were calculated by dividing each normalized expression value with the mean of the respective control group and values were log10 transformed for statistical analysis. ROUT method [
88] (combines a robust nonlinear regression with an outlier identification method based on the false discovery rate) was used to detect outliers. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was performed for between-group comparisons. If Bartlett’s test indicated heterogeneity of variances, the Kruskal–Wallis one-way analysis of variance by ranks was performed followed by Dunn’s test. If there were only two groups, unpaired Student’s t-test was used for comparison. The null hypothesis was rejected if
p < 0.05. Statistical analysis was performed using GraphPad Prism6 (GraphPad Software Inc., San Diego, CA, USA).

 ,
, 





{kind=link}
{kind=link}
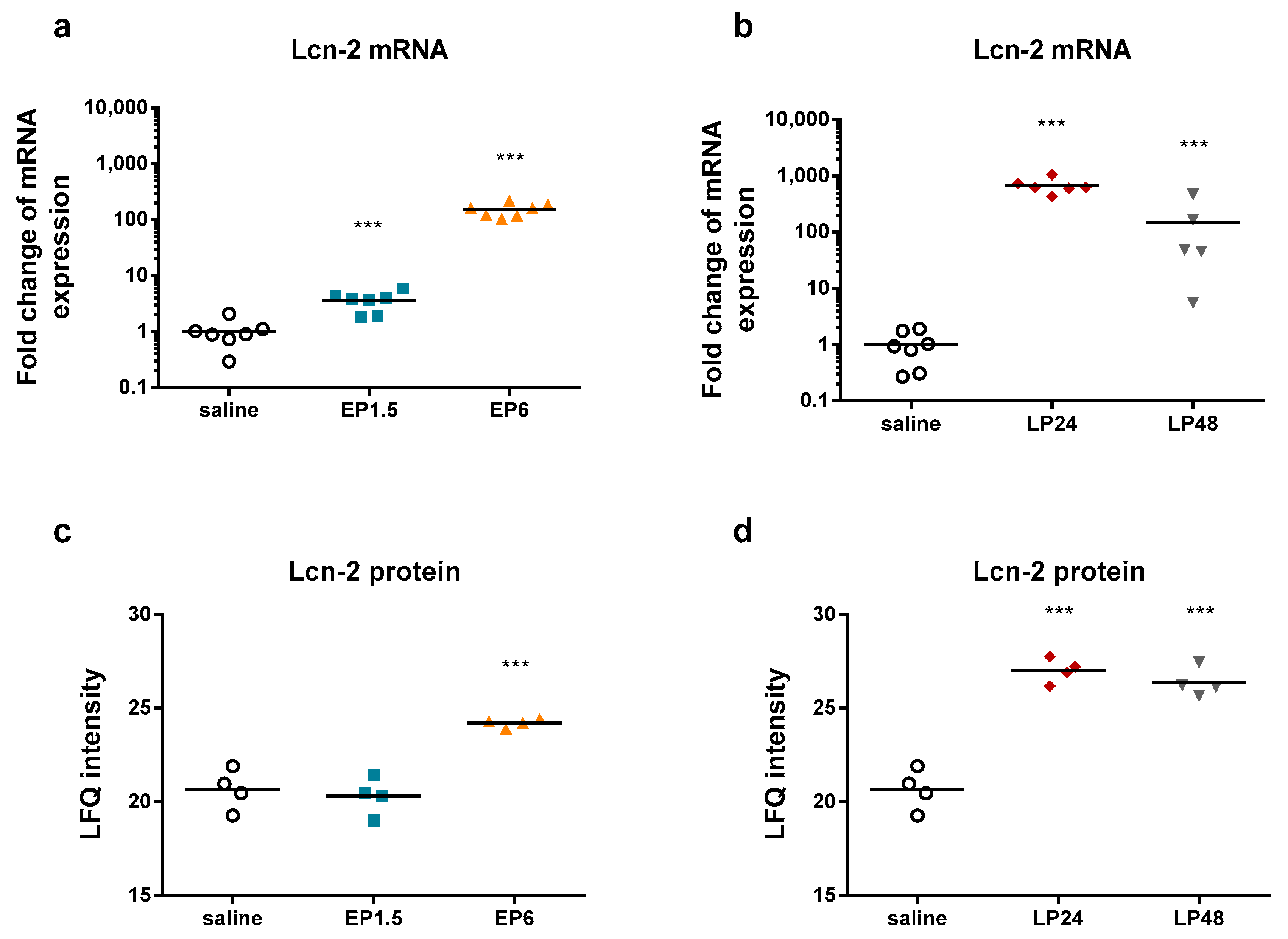{kind=link}
{kind=link}
{kind=link}
{kind=link}