Ferroptosis in Liver Diseases: An Overview


Abstract
:1. Introduction
2. Discovery of Ferroptosis
3. An Overview of Ferroptosis
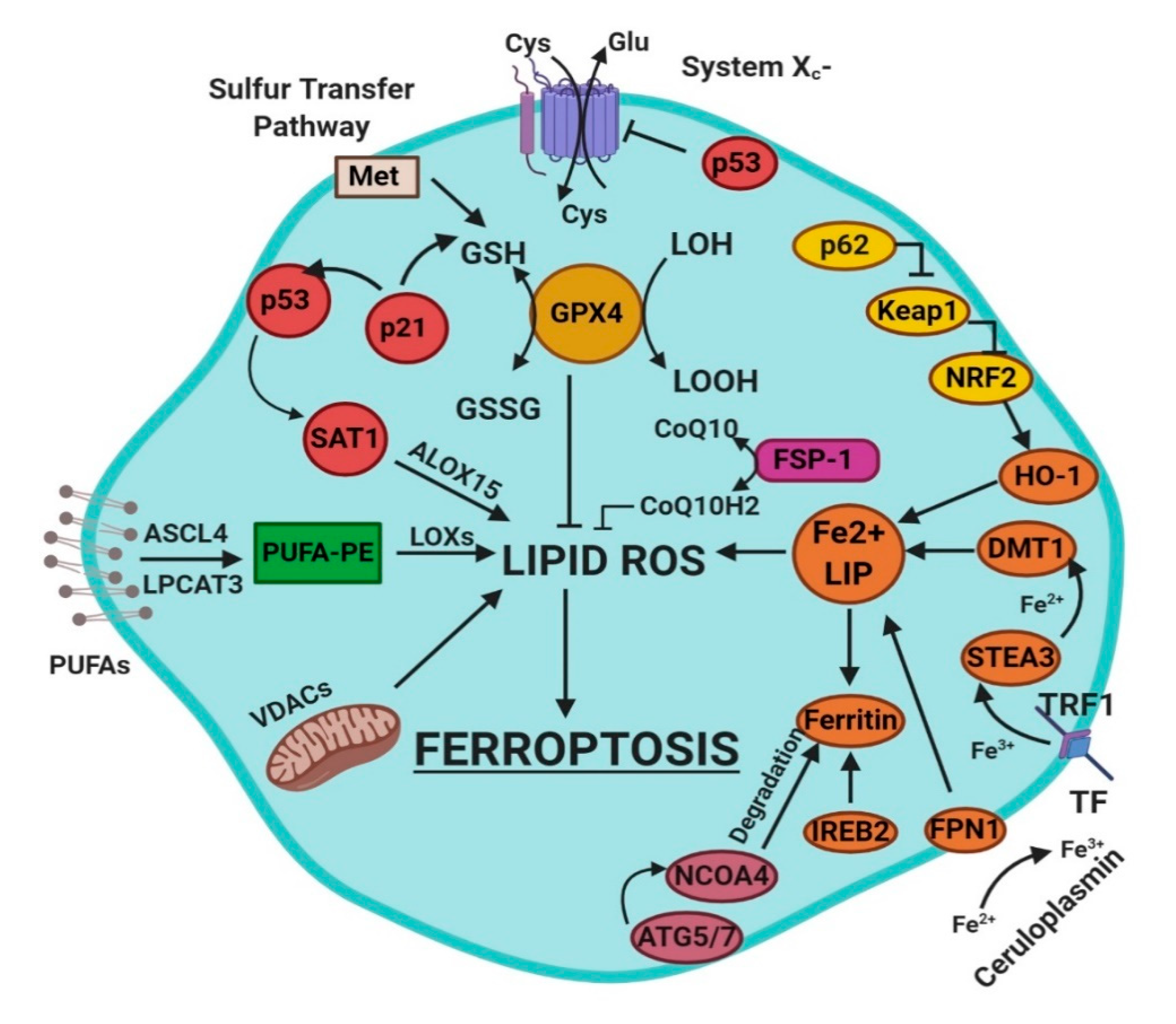
4. Mechanisms of Ferroptosis
- glutathione/glutathione peroxidase 4 (GSH/GPX4) pathway, inhibition of system Xc−, sulfur transfer pathway, and p53 regulatory axis;
- iron metabolism with the regulation of autophagy protein 5 and 7 (ATG5-ATG7) and nuclear receptor coactivator 4 (NCOA4) pathway and iron-responsive element-binding protein 2 (IREB2) related to ferritin metabolism, and the p62-Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor (Nrf2) regulatory pathways [3]; and
- lipid metabolism pathways as p53, arachidonate lipoxygenase 15 (ALOX15), acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3) [3].
4.1. Radical Oxygen Species (ROS)
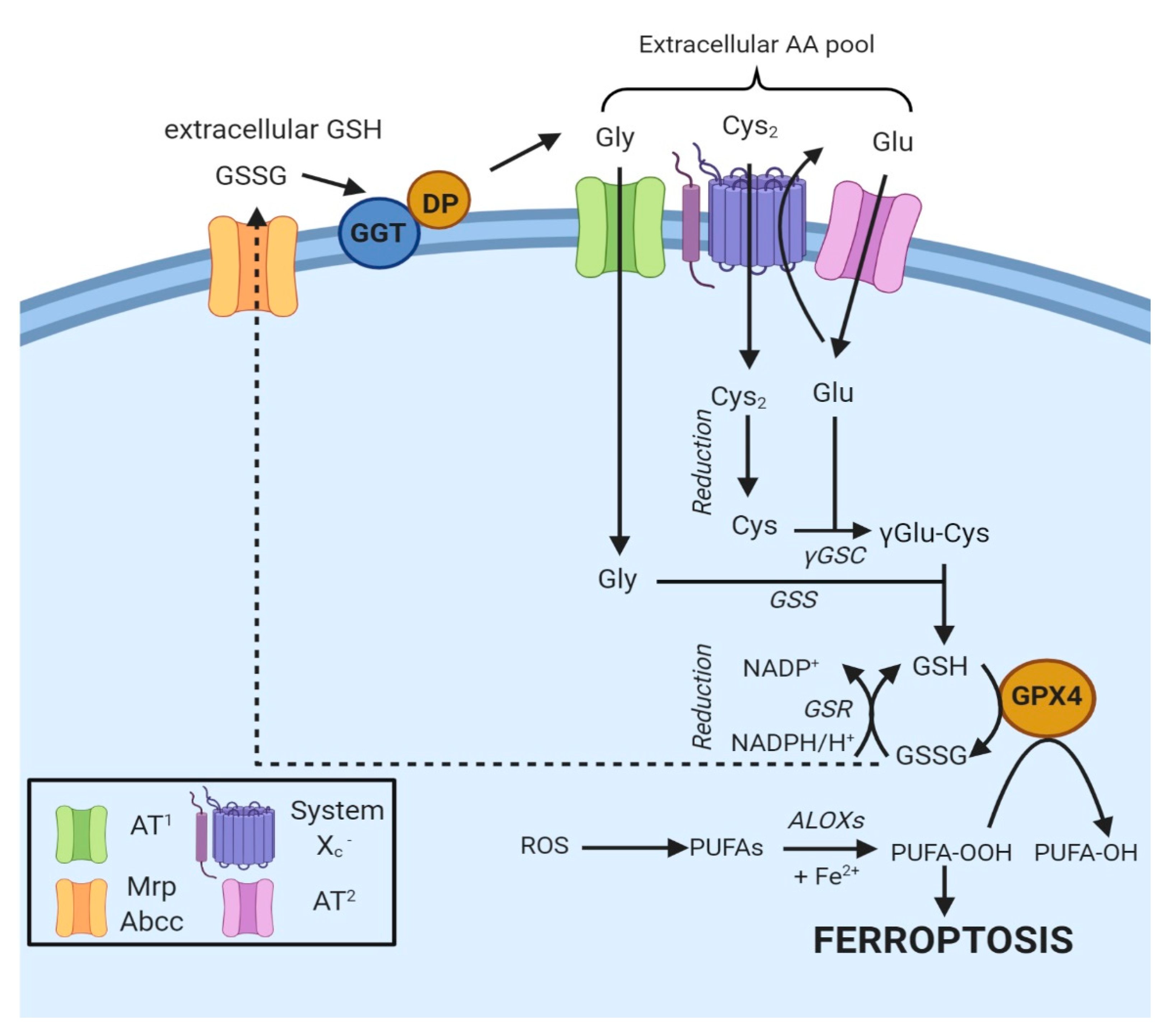
4.2. Regulation of Ferroptosis via Cysteine-Glutathione Redox Axis
4.2.1. Biosynthesis of Glutathione
4.2.2. Uptake of Cysteine
4.2.3. System Xc−
4.2.4. Cysteine Synthesis via the Transsulfuration Pathway
4.3. The Enzyme Glutathione Peroxidase 4 and Ferroptosis
4.4. Lipid Metabolism and Ferroptosis
4.4.1. Role of Lipoxygenases
4.4.2. Lipid Peroxidation
4.4.3. Where Does Lipid Peroxidation Take Place?
4.5. Iron Metabolism and Ferroptosis
4.6. p53-Mediated Ferroptosis
4.7. Ferroptosis and the Keap1–Nrf2 Pathway
4.8. Other Related Signaling Pathways
5. Crosstalk between Autophagy and Ferroptosis
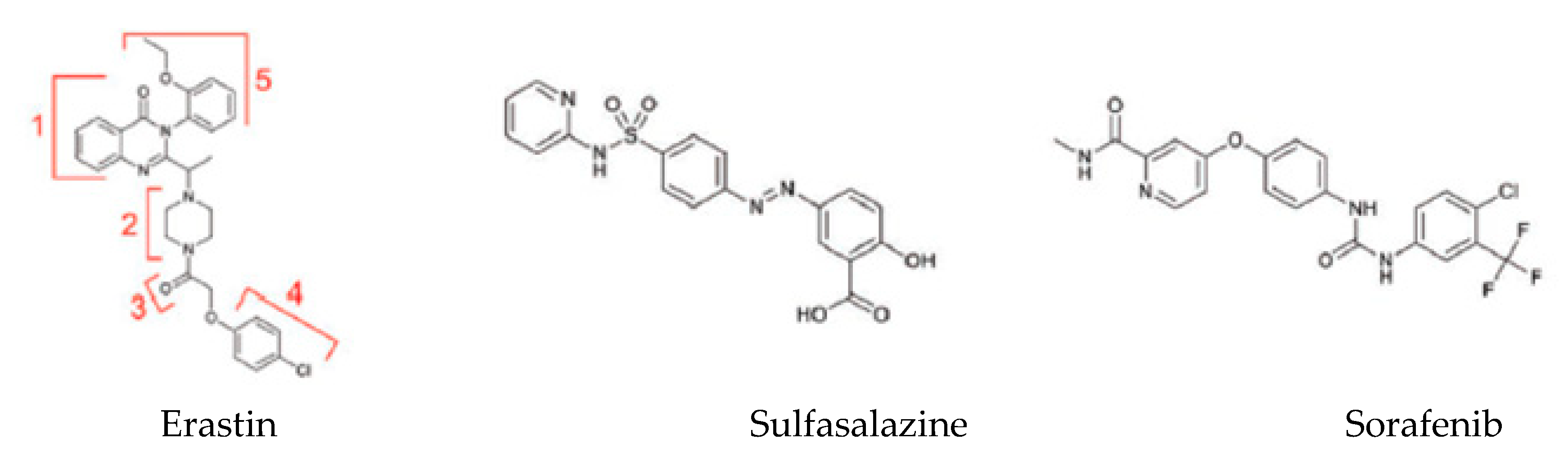
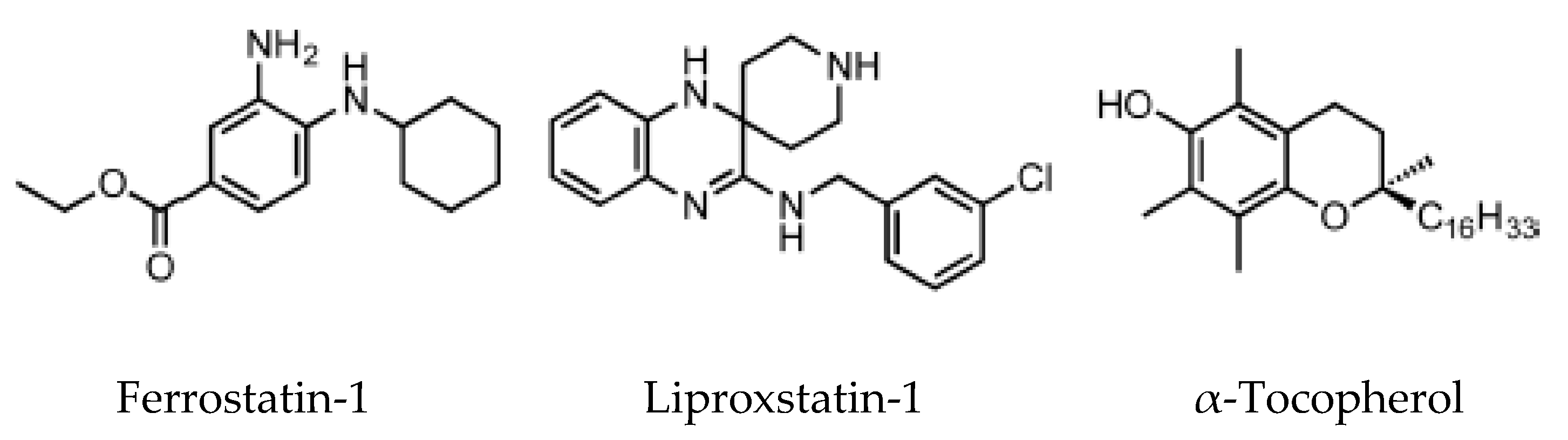
6. Pharmacological Modulation of Ferroptosis
6.1. Ferroptosis Inducers
6.2. Ferroptosis Inhibitors
7. Ferroptosis in Liver Diseases
7.1. Ferroptosis and Drug-Induced Liver Injury
7.2. Ferroptosis and Ischemia–Reperfusion Injury (IRI)
7.3. Chronic Liver Diseases (CLD)
7.4. Hepatocellular Carcinoma (HCC)
8. Conclusion and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-noenal |
| AA | Arachidonic acid |
| ACC | Acetyl-CoA carboxylase |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| ALD | Acute liver disease |
| ALOXs | Arachidonate lipoxygenases |
| AREs | Antioxidant response elements |
| ATG5-7 | Aumiddlehagy protein 5 and 7 |
| bZIP | Basic leucine zipper |
| CBS | Cystathionine β-synthase |
| CDKN1A/p21 | Cyclin-dependent kinase inhibitor 1A |
| CISD1 | CDGSH iron sulfur domain 1 |
| CLD | Chronic liver disease |
| CoQ10 | Coenzyme Q10 |
| CRC | Colorectal cancer |
| DAMPs | Damage-associated molecular patterns |
| DCYTB | Duodenal cytochrome B reductase |
| DFO | Deferoxamine |
| DILI | Drug-induced liver injury |
| DMT1 | Divalent metal transporter 1 |
| DPP4 | Dipeptidyl-peptidase 4 |
| ER | Endoplasmic reticulum |
| Fe2+ | Ferrous iron |
| Fe3+ | Ferric iron |
| FECH | Ferrochelatase |
| Fer-1 | Ferrostatin-1 |
| FPN | Ferroportin |
| FSP1 | Ferroptosis suppressor protein 1 |
| FTH1 | Ferritin heavy chain |
| FTL | Ferritin light chain |
| GGT | γ-Glutamyl transpeptidase |
| GLC | γ-Glu-Cys-ligase |
| GPX4 | Glutathione peroxidase 4 |
| GSR | Glutathione reductase |
| GSH | Glutathione |
| GSS | Glutathione synthetase |
| HATs | Heterodimeric amino acid transporters |
| HCC | Hepatocellular carcinoma |
| HCP-1 | Heme carrier protein-1 |
| HO-1 | Heme oxygenase-1 |
| HSC | Hemamiddleoietic Stem Cell |
| HSPs | Heat shock proteins |
| HSPB1 | Heat shock factor-binding protein 1 |
| IREB2 | Iron-responsive element-binding protein 2 |
| IRI | Ischemia–reperfusion injury |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LC3-I/II | Microtubule-associated protein light chain 3 |
| LIP | Labile iron pool |
| Lip-1 | Liproxstatin-1 |
| LPCAT3 | Lysophosphatidylcholine acyltransferase 3 |
| MAPKs | Mitogen-activated protein kinases |
| MDA | Malondialdehyde |
| MRP/ABCC | Multidrug resistance-associated proteins |
| MT-1G | Metallothionein-1G |
| NAFLD | Non-alcoholic liver disease |
| NAPQI | N-acetyl-p-benzoquinone imine |
| NASH | Non-alcoholic steatohepatitis |
| NCOA4 | Nuclear receptor coactivator 4 |
| Nrf2 | Nuclear factor erythroid 2-related factor |
| OMM | Outer mitochondrial membrane |
| OXPHOS | Oxidative phosphorylation |
| PARP1 | Poly [ADP-ribose] polymerase 1 |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PEPT1-2 | Proton-coupled oligopeptide transporter family members 1 and 2 |
| PKC | Protein kinase C |
| PS | Phosphatidylserine |
| PUFAs | Polyunsaturated fatty chains |
| Rb | Retinoblastoma |
| RCD | Regulated cell death |
| ROS | Reactive oxygen species |
| RPTECs | Renal proximal tubular epithelial cells |
| RSL3/5 | Ras-selective lethal small molecule 3/5 |
| RTA | Radical-trapping antioxidant |
| SAS | Sulfasalazine |
| SAT1 | Spermidine/spermine N1-acetyltransferase 1 |
| SOD3 | Superoxide dismutase 3 |
| STEAP3 | Six-transmembrane epithelial antigen of prostate 3 |
| Tf | Transferrin |
| TGFβ | Transforming growth factor beta |
| TRF1 | Transferrin receptor protein 1 |
| TXN | Thioredoxin |
| TXNRD1 | Thioredoxin reductase 1 |
| VDACs | Voltage-dependent anion channels |
References
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison: Iron and Ferroptosis’Ferroptosis’. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell ‘Death’. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Fujii, J.; Kobayashi, S.; Homma, T. Regulation of Ferroptosis Through the Cysteine-Glutathione Redox Axis. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 197–213. [Google Scholar]
- Ran, Q.; Mozolewska, P. Gpx4 and Ferroptosis. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 99–109. [Google Scholar]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Tang, D. (Ed.) Ferroptosis in Health and Disease; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free. Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x c − : Cystine supplier and beyond. Amino Acids 2011, 42, 231–246. [Google Scholar] [CrossRef]
- Newstead, S. Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 488–499. [Google Scholar] [CrossRef] [Green Version]
- Bannai, S.; Kitamura, E. Transport Interactionof t-Cystine and L-Glutamatein Human Diploid Fibroblasts in Culture. Biol. Chem. 1980, 255, 2372–2376. [Google Scholar]
- Lewerenz, J.; Hewett, S.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System xc− in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxidants Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kang, E.S.; Kobayashi, S.; Homma, T.; Sato, H.; Seo, H.G.; Fujii, J. The viability of primary hepatocytes is maintained under a low cysteine-glutathione redox state with a marked elevation in ophthalmic acid production. Exp. Cell Res. 2017, 361, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2015, 23, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Yant, L.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free. Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Scheerer, P.; Borchert, A.; Kraus, N.; Wessner, H.; Gerth, C.; Höhne, W.; Kuhn, H. Structural Basis for Catalytic Activity and Enzyme Polymerization of Phospholipid Hydroperoxide Glutathione Peroxidase-4 (GPx4)†,‡,§. Biochemistry 2007, 46, 9041–9049. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Tarangelo, A.; Dixon, S.J. Lipid Metabolism and ‘Ferroptosis’. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 1–26. [Google Scholar]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Daher, R.; Manceau, H.; Karim, Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. La Presse Médicale 2017, 46, e272–e278. [Google Scholar] [CrossRef]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, T.; Miki, A.; Nagasawa, H. Organelle-specific analysis of labile Fe(ii) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 2019, 11, 111–117. [Google Scholar] [CrossRef]
- Wang, S.-J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Copple, I.M. The Keap1–Nrf2 Cell Defense Pathway–A Promising Therapeutic Target? Adv. Pharmacol. 2012, 63, 43–79. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Liakopoulos, V.; Stefanidis, I. The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death. Biology 2019, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.; Bolisetty, S. Ferritins in Kidney Disease. Semin. Nephrol. 2020, 40, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta BBA Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 0168827820302105. [Google Scholar] [CrossRef]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef] [Green Version]
- Kullak-Ublick, G.A.; Andrade, R.J.; Merz, M.; End, P.; Benesic, A.; Gerbes, A.L.; Aithal, G.P. Drug-induced liver injury: Recent advances in diagnosis and risk assessment. Gut 2017, 66, 1154–1164. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, A. Ferroptosis in Liver Disease. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 239–248. [Google Scholar]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Saidi, R.F.; Kenari, S.K.H. Liver Ischemia/Reperfusion Injury: An Overview. J. Investig. Surg. 2014, 27, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Afonso, M.B.; Castro, R.E.; Rodrigues, C.M.P. Processes exacerbating apoptosis in non-alcoholic steatohepatitis. Clin. Sci. 2019, 133, 2245–2264. [Google Scholar] [CrossRef]
- Loguercio, C.; De Girolamo, V.; De Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Di Marino, M.P.; et al. Non-alcoholic fatty liver disease in an area of southern Italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; LaVine, J.E.; Tonascia, J.; Ünalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. New Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Bonkovsky, H.L.; Jawaid, Q.; Tortorelli, K.; LeClair, P.; Cobb, J.; Lambrecht, R.W.; Banner, B.F. Non-alcoholic steatohepatitis and iron: Increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 421–429. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Ye, T.J.; Bonavita, G.; Daniels, M.; Kainrad, N.; Jogasuria, A.; You, M. Adipose-Specific Lipin-1 Overexpression Renders Hepatic Ferroptosis and Exacerbates Alcoholic Steatohepatitis in Mice. Hepatol. Commun. 2019, 3, 656–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Caldas, C.; Santos, A.G.; Silva-Gomes, S.; Santos-Gonçalves, A.; Martins, M.J.; Porto, G.; Lopes, J.M. Genetic disruption of NRF2 promotes the development of necroinflammation and liver fibrosis in a mouse model of HFE-hereditary hemochromatosis. Redox Biol. 2017, 11, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337. [Google Scholar] [CrossRef]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; Francois, C.; Chatelain, D.; DeBuysscher, V.; Barbare, J.-C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef]
- Ou, W.; Mulik, R.S.; Anwar, A.; McDonald, J.G.; He, X.; Corbin, I.R. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free. Radic. Biol. Med. 2017, 112, 597–607. [Google Scholar] [CrossRef]
- Bai, T.; Wang, S.; Zhao, Y.; Zhu, R.; Wang, W.; Sun, Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 491, 919–925. [Google Scholar] [CrossRef]
- Bebber, C.M.; Müller, F.; Clemente, L.P.; Weber, J.; Von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Death | Morphological Features | Biochemical Features |
|---|---|---|
| Ferroptosis | No rupture of the plasma membrane [3] Rounding up of the cell [3] Small mitochondria, outer mitochondrial rupture, reduction of the cristae [3] Normal nuclear size and no chromatin condensation [3] | Iron and ROS overload Activation of MAPKs Inhibition of system Xc− and decreased cystine uptake GSH depletion Release of arachidonic acid mediators [3] |
| Apoptosis | Plasma membrane blebbing [3] Rounding up of the cell [3] Pseudopod retraction and reduction of cellular and nuclear volume [3] Nuclear fragmentation, chromatin condensation Formation of apoptotic bodies [3] No significant changes in mitochondrial structure [3] | Activation of caspases Oligonucleosomal DNA fragmentation PS exposure [3] |
| Necroptosis | Rupture of the plasma membrane [3] Cytoplasmic swelling [3] Moderate chromatin condensation [5] Spillage of cellular constituents into microenvironment [3] | Decrease in ATP level Release DAMPs PARP1 hyperactivation [3] |
| Autophagy | Lack of change in the plasma membrane [3] Accumulation of autophagic vacuoles [3] Lack of chromatin condensation [5] Formation of double-membraned autolysosomes, including macroautophagy, microautophagy and chaperone-mediated autophagy [3] | LC3-I to LC3-II conversion [3] Substrate degradation [5] |
| Class | Class Characteristics | Impact on Ferroptosis | Compound Examples | Suitable for In Vivo Use |
|---|---|---|---|---|
| Class 1 | Inhibition of system Xc− | Prevention of cystine import, GSH depletion, loss of GPX4 activity | Erastin, sulfasalazine, sorafenib | Sorafenib, sulfasalazine |
| Class 2 | Direct inhibition of GPX4 | Covalent interaction with GPX4 and inhibition of the enzyme | RSL3, RSL5 | No |
| Class 3 | Depletion of GPX4 protein and CoQ10 | Depletion of GPX4 and CoQ10 | FIN56 | Unknown |
| Class 4 | Induction of lipid peroxidation | Oxidation of iron drives lipid peroxidation and indirect inactivation of GPX4 | FINO2 | Unknown |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capelletti, M.M.; Manceau, H.; Puy, H.; Peoc’h, K. Ferroptosis in Liver Diseases: An Overview. Int. J. Mol. Sci. 2020, 21, 4908. https://doi.org/10.3390/ijms21144908
Capelletti MM, Manceau H, Puy H, Peoc’h K. Ferroptosis in Liver Diseases: An Overview. International Journal of Molecular Sciences. 2020; 21(14):4908. https://doi.org/10.3390/ijms21144908
Chicago/Turabian StyleCapelletti, Martina Maria, Hana Manceau, Hervé Puy, and Katell Peoc’h. 2020. "Ferroptosis in Liver Diseases: An Overview" International Journal of Molecular Sciences 21, no. 14: 4908. https://doi.org/10.3390/ijms21144908