The Regulatory Network of CMPG1-V in Wheat–Blumeria graminis f. sp. tritici Interaction Revealed by Temporal Profiling Using RNA-Seq

Abstract
:1. Introduction
2. Results
2.1. Global Profiling of CMPG1-VOE in Response to Bgt Infection
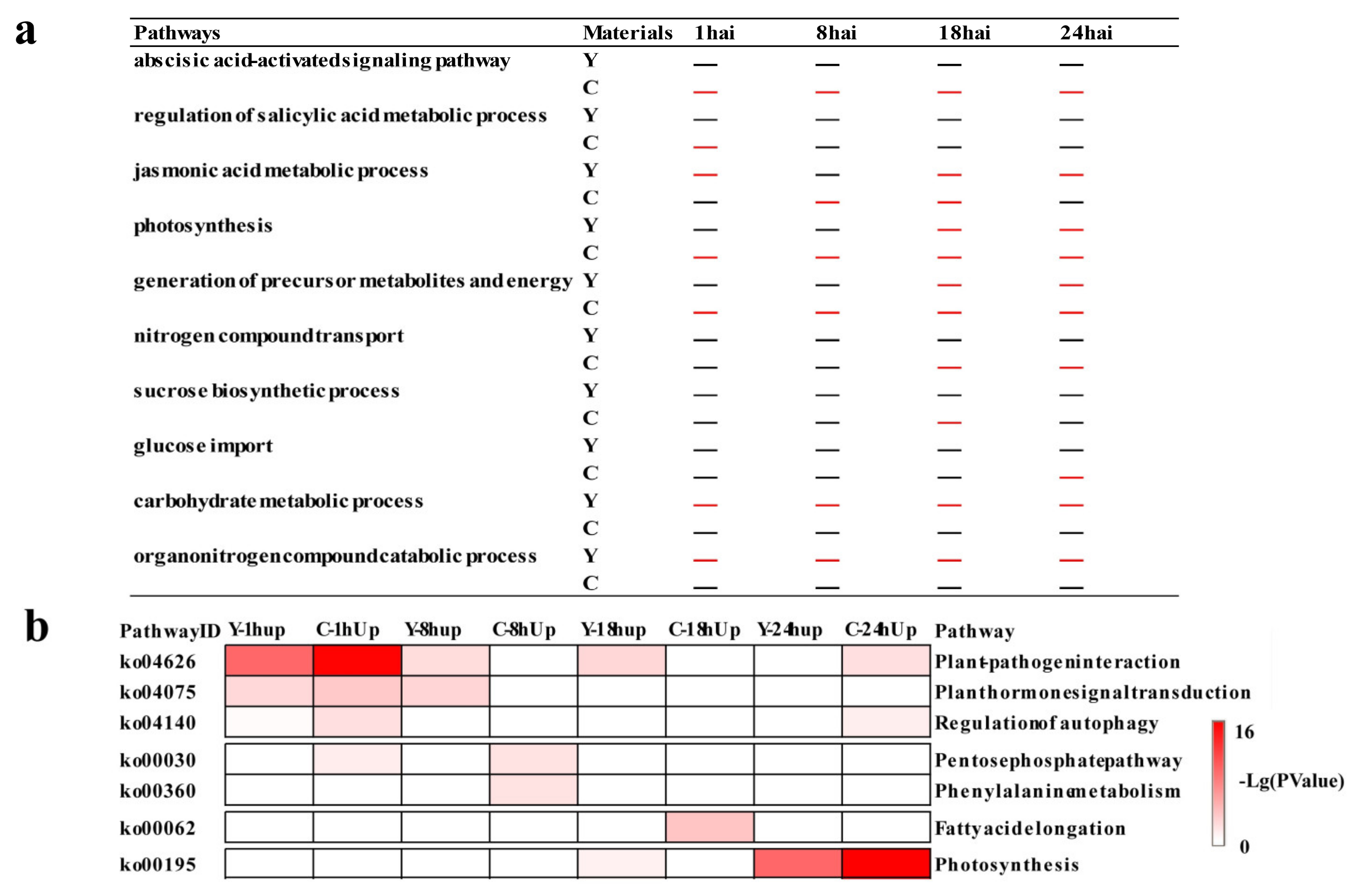
2.2. Functional Categories Enriched in CMPG1-VOE and Yangmai 158 in Response to Bgt Infection
2.3. Temporal Specificity of CMPG1-VOE in Response to Bgt Infection
2.4. Phytohormone Signaling Was Rapidly Reprogrammed in CMPG1-VOE after Bgt Infection
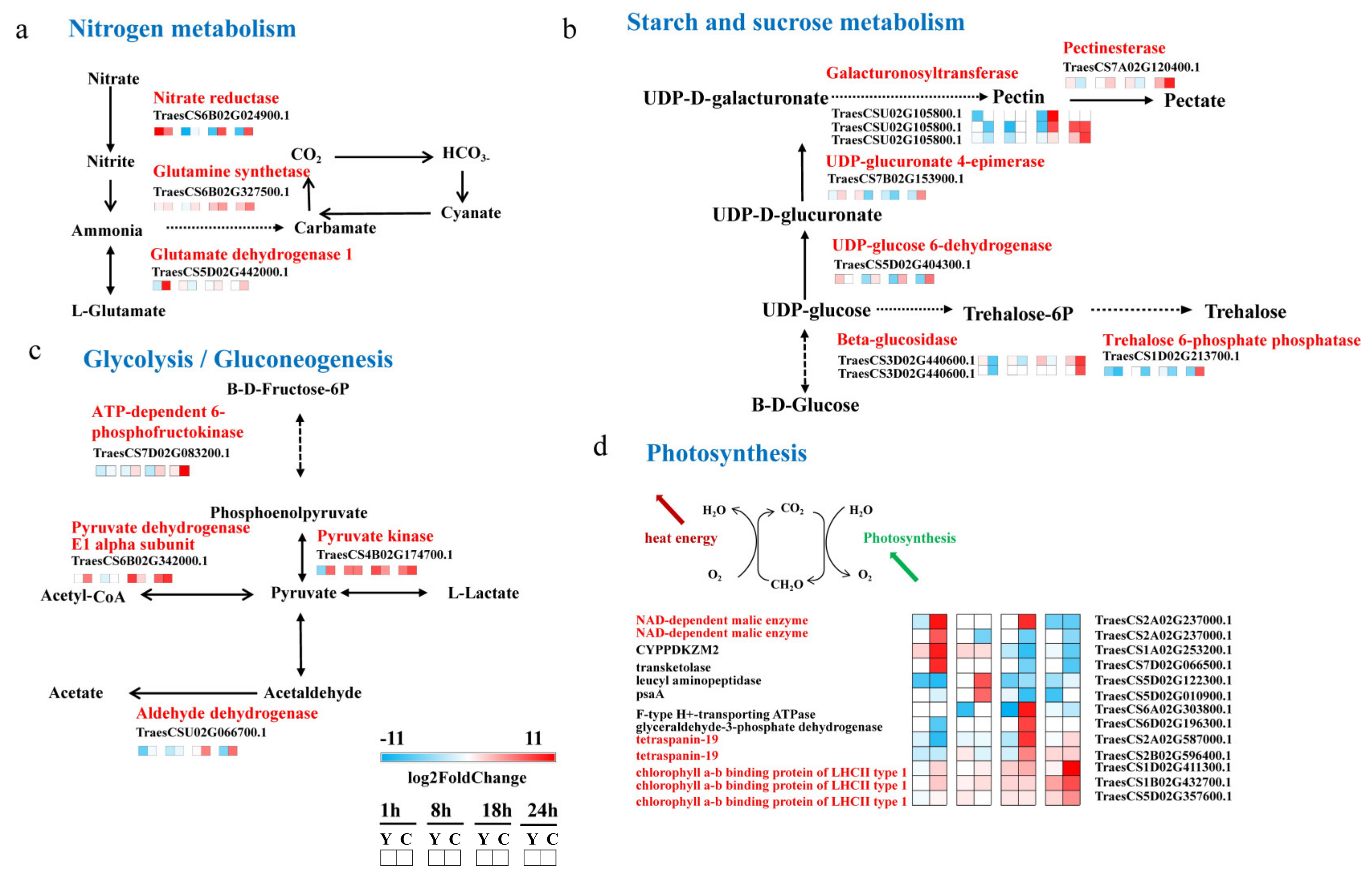
2.5. CMPG1-V Activates Conspicuous Energy Metabolic Signaling during Bgt Infection
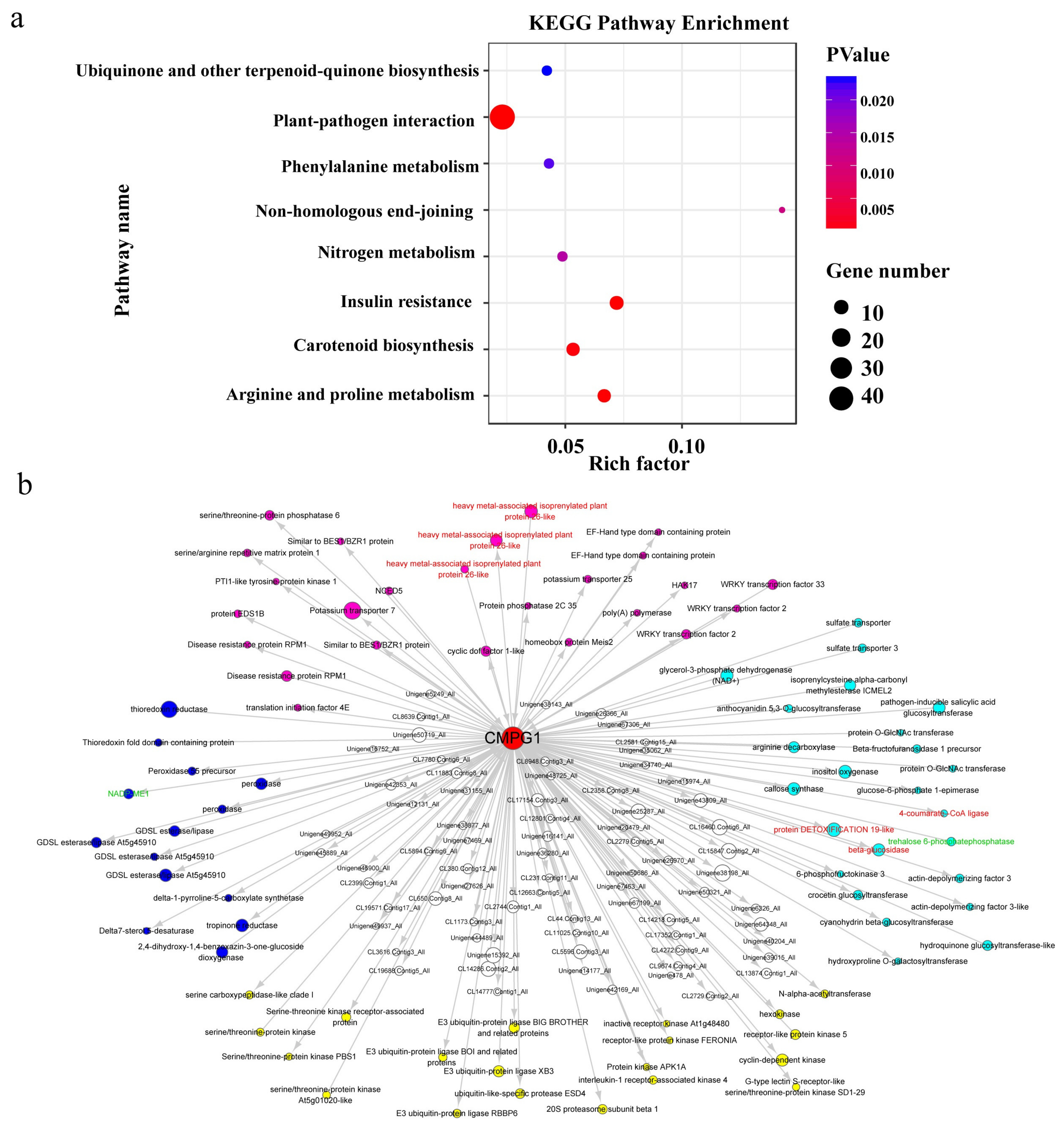
2.6. Weighted Gene Co-Expression Network (WGCNA) of Pm Resistance Regulated by CMPG1-V
2.7. The Chromosomal Distribution of Candidate Genes
3. Discussion
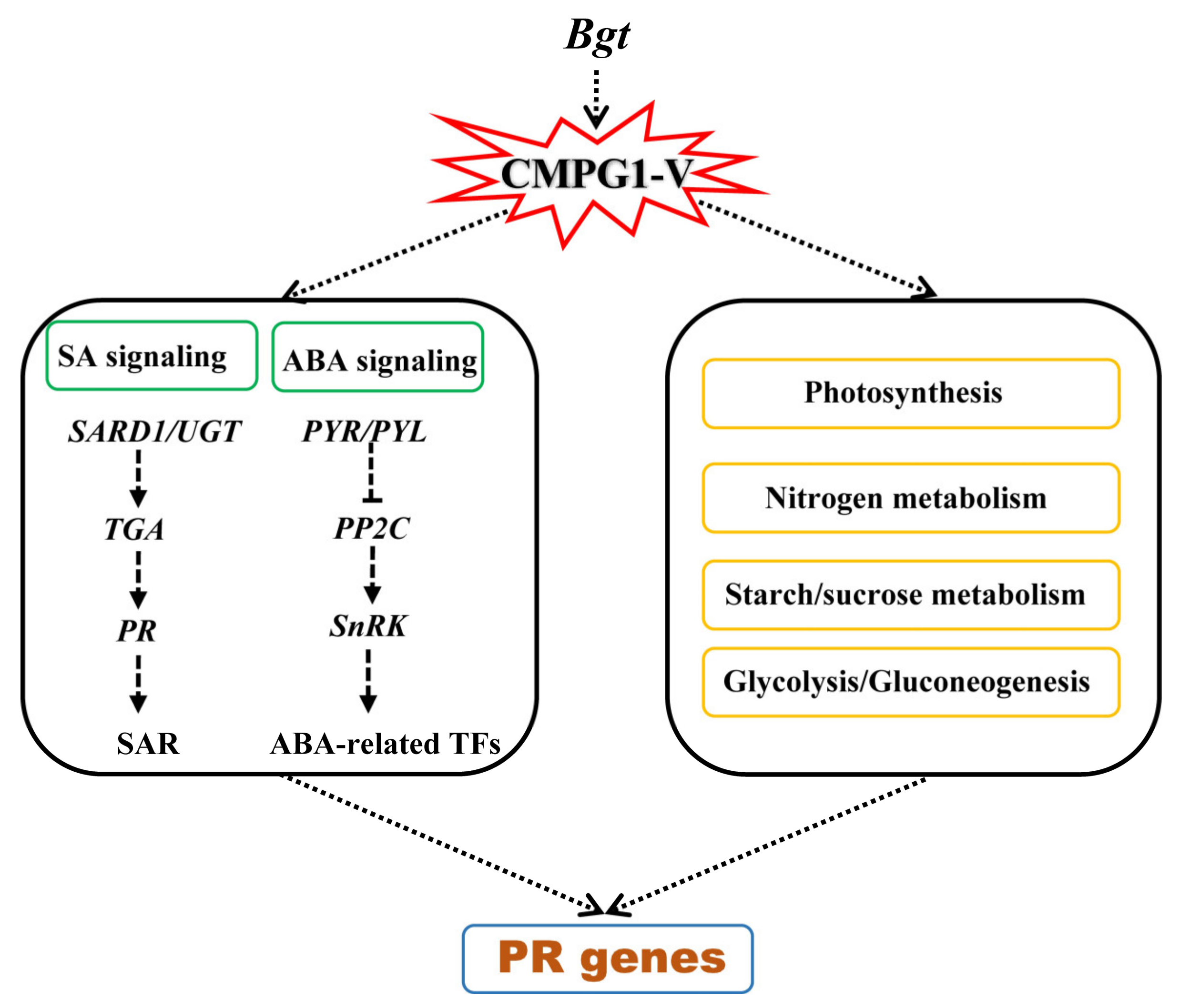
3.1. Phytohormone SA and ABA Play Important Roles in CMPG1-V-Associated Powdery Mildew Resistance
3.2. CMPG1-V Reprogrammed Starch and Sucrose Metabolism/Photosynthesis in Response to Bgt Infection
3.3. CMPG1-V Exerts Its Distinct Defense Response by Reprogramming a Specific Network
4. Materials and Methods
4.1. Plant Material and Fungal Isolates
4.2. RNA-Seq Library Construction and Sequencing
4.3. Sequencing Reads Filtering and De Novo Assembly
4.4. Unigene Functional Annotation and Expression Calculation
4.5. Detection and Analysis of Differentially Expressed Gene
4.6. Weighted Gene Co-Expression Network Analysis and Visualization
4.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4CL | 4-coumarate-CoA ligase |
| ABA | abscisic acid |
| AGT | appressorial germ tube |
| ALDH | aldehyde dehydrogenase |
| Bgt | Blumeria graminis f. sp. tritici |
| bp | base pair |
| BSR | broad-spectrum resistance |
| DEG | differentially expressed gene |
| ET | ethylene |
| GAE | UDP-glucuronate 4-epimeras |
| GAUT | galacturonosyltransferase |
| GDH1 | glutamate dehydrogenase 1 |
| GLUC | beta-glucosidase |
| GO | Gene Ontology |
| GS | glutamine synthetase |
| GT | anthocyanidin 5,3-O-glucosyltransferase |
| HA | hyphal appressoria |
| JA | jasmonic acid |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LHCII | chlorophyll a-b binding protein |
| NAD-ME | NAD-dependent malic enzyme |
| NR | nitrate reductase |
| PDHA | pyruvate dehydrogenase E1 alpha subunit |
| PFK3 | 6-phosphofructokinase 3 |
| pfkA | ATP-dependent 6-phosphofructokinase |
| PGT | primary germ tube |
| PK | pyruvate kinase |
| Pm | powdery mildew |
| PR | pathogenesis-related gene |
| PUB | plant U-box type E3 ubiquitin ligases |
| RNA-seq | RNA-sequencing |
| SA | salicylic acid |
| SARD1 | SAR DEFICIENT1 |
| SnRK | SNF1-related protein kinase |
| TPP | trehalose 6-phosphatephosphatase |
| TSPAN | tetraspanin-19 |
| UGDH | UDP-glucose 6-dehydrogenase |
| UGT | UDP-glycosyltransferase |
| WGCNA | Weighted Gene Co-Expression Network Analysis |
References
- Berens, M.L.; Berry, H.M.; Mine, A.; Argueso, C.T.; Tsuda, K. Evolution of hormone signaling networks in plant defense. Annu. Rev. Phytopathol. 2017, 55, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Mithoe, S.C.; Menke, F.L. Regulation of pattern recognition receptor signalling by phosphorylation and ubiquitination. Curr. Opin. Plant. Biol. 2018, 45, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Thines, B.; Katsir, L.; Melotto, M.; Niu, Y.; Mandaokar, A.; Liu, G.; Nomura, K.; He, S.Y.; Howe, G.A.; Browse, J. JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature 2007, 448, 661–665. [Google Scholar] [CrossRef]
- Pauwels, L.; Ritter, A.; Goossens, J.; Durand, A.N.; Liu, H.; Gu, Y.; Geerinck, J.; Boter, M.; Vanden Bossche, R.; De Clercq, R.; et al. The RING E3 ligase KEEP ON GOING modulates JASMONATE ZIMDOMAIN12 stability. Plant. Physiol. 2015, 169, 1405–1417. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Hu, T.; Bao, M.; Cao, L.; Zhang, H.; Song, F.; Xie, Q.; Zhou, X. Tobacco RING E3 ligase NtRFP1 mediates ubiquitination and proteasomal degradation of a geminivirus-encoded betaC1. Mol. Plant. 2016, 9, 911–925. [Google Scholar] [CrossRef] [Green Version]
- Boyle, P.; Le Su, E.; Rochon, A.; Shearer, H.L.; Murmu, J.; Chu, J.Y.; Fobert, P.R.; Després, C. The BTB/POZ domain of the Arabidopsis disease resistance protein NPR1 interacts with the repression domain of TGA2 to negate its function. Plant. Cell 2009, 21, 3700–3713. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, H.; Miao, M.; Tang, X.; Giovannoni, J.; Xiao, F.; Liu, Y. The tomato UV-damaged DNA-binding protein-1 (DDB1) is implicated in pathogenesis related (PR) gene expression and resistance to Agrobacterium tumefaciens. Mol. Plant. Pathol. 2012, 13, 123–134. [Google Scholar] [CrossRef]
- Zhang, L.; Du, L.; Shen, C.; Yang, Y.; Poovaiah, B.W. Regulation of plant immunity through ubiquitin-mediated modulation of Ca2+-calmodulin-AtSR1/CAMTA3 signaling. Plant J. 2014, 78, 269–281. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, Y.; Zhang, H.; Li, D.; Zheng, Z.; Song, F. Overexpression of a rice defense-related F-box protein gene OsDRF1 in tobacco improves disease resistance through potentiation of defense gene expression. Physiol. Plant. 2008, 134, 440–452. [Google Scholar] [CrossRef]
- Lin, S.S.; Martin, R.; Mongrand, S.; Vandenabeele, S.; Chen, K.C.; Jang, I.C.; Chua, N.H. RING1 E3 ligase localizes to plasma membrane lipid rafts to trigger FB1-induced programmed cell death in Arabidopsis. Plant J. 2008, 56, 550–561. [Google Scholar] [CrossRef]
- Lee, D.H.; Choi, H.W.; Hwang, B.K. The pepper E3 ubiquitin ligase RING1 gene, CaRING1, is required for cell death and the salicylic acid-dependent defense response. Plant Physiol. 2011, 156, 2011–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Maekawa, S.; Yasuda, S.; Domeki, Y.; Sueyoshi, K.; Fujiwara, M.; Fukao, Y.; Goto, D.B.; Yamaguchi, J. Identification of 14-3-3 proteins as a target of ATL31 ubiquitin ligase, a regulator of the C/N response in Arabidopsis. Plant J. 2011, 68, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Maekawa, S.; Yasuda, S.; Sonoda, Y.; Katoh, E.; Ichikawa, T.; Nakazawa, M.; Seki, M.; Shinozaki, K.; Matsui, M.; et al. CNI1/ATL31, a RING-type ubiquitin ligase that functions in the carbon/nitrogen response for growth phase transition in Arabidopsis seedlings. Plant J. 2009, 60, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Sato, T.; Asada, Y.; Yasuda, S.; Yoshida, M.; Chiba, Y.; Yamaguchi, J. The Arabidopsis ubiquitin ligases ATL31 and ATL6 control the defense response as well as the carbon/nitrogen response. Plant. Mol. Biol. 2012, 79, 217–227. [Google Scholar] [CrossRef]
- Godfray, H.C.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Menardo, F.; Praz, C.R.; Wyder, S.; Ben-David, R.; Bourras, S.; Matsumae, H.; McNally, K.E.; Parlange, F.; Riba, A.; Roffler, S.; et al. Hybridization of powdery mildew strains gives rise to pathogens on novel agricultural crop species. Nat. Genet. 2016, 48, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, Y.; Fei, F.; Wang, Z.; Wang, W.; Cao, A.; Liu, Y.; Han, S.; Xing, L.; Wang, H.; et al. E3 ubiquitin ligase gene CMPG1-V from Haynaldia villosa L. contributes to powdery mildew resistance in common wheat (Triticum aestivum L.). Plant. J. 2015, 84, 154–168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Henderson, C.; Perfect, E.; Carver, T.L.; Thomas, B.J.; Skamnioti, P.; Gurr, S.J. Of genes and genomes, needles and haystacks: Blumeria graminis and functionality. Mol. Plant. Pathol. 2005, 6, 561–575. [Google Scholar] [CrossRef]
- Lalonde, S.; Sero, A.; Pratelli, R.; Pilot, G.; Chen, J.; Sardi, M.; Parsa, S.A.; Kim, D.Y.; Acharya, B.R.; Stein, E.V.; et al. A membrane protein/signaling protein interaction network for Arabidopsis version AMPv2. Front. Physiol. 2010, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, S.; Chen, F.; Chen, H.; Wang, J.; McCall, C.; Xiong, Y.; Deng, X.W. Arabidopsis DDB1-CUL4 ASSOCIATED FACTOR1 forms a nuclear E3 ubiquitin ligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant. Cell. 2008, 6, 1437–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benschop, J.J.; Mohammed, S.; O’Flaherty, M.; Heck, A.J.; Slijper, M.; Menke, F.L. Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell Proteomics. 2007, 6, 1198–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, A.R.; Ashfield, T.; Innes, R.W. Pseudomonas syringae effector AvrPphB suppresses AvrB-induced activation of RPM1 but not AvrRpm1-induced activation. Mol. Plant. Microbe Interact. 2015, 28, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Parsons, H.T.; Christiansen, K.; Knierim, B.; Carroll, A.; Ito, J.; Batth, S.T.; Smith-Moritz, A.M.; Morrison, S.; McInerney, P.; Hadi, M.Z.; et al. Isolation and proteomic characterization of the Arabidopsis Golgi defines functional and novel components involved in plant cell wall biosynthesis. Plant. Physiol. 2012, 159, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; Liu, M.J.; Kita, D.; Jordan, S.S.; Yeh, F.L.; Yvon, R.; Carpenter, H.; Federico, A.N.; Garcia-Valencia, L.E.; Eyles, S.J.; et al. FERONIA controls pectin- and nitric oxide-mediated male-female interaction. Nature 2020, 579, 561–566. [Google Scholar] [CrossRef]
- Tarutani, Y.; Sasaki, A.; Yasuda, M.; Nakashita, H.; Yoshida, S.; Yamaguchi, I.; Suzuk, Y. Identification of three clones which commonly interact with the kinase domains of highly homologous two receptor-like kinases, RLK902 and RKL1. Biosci. Biotechnol. Biochem. 2004, 68, 2581–2587. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Fukuzawa, N.; Hyodo, A.; Kim, H.; Mashiyama, S.; Ogihara, T.; Yoshioka, H.; Matsuura, H.; Masuta, C.; Matsumura, T.; et al. Role of salicylic acid glucosyltransferase in balancing growth and defence for optimum plant fitness. Mol. Plant. Pathol. 2020, 21, 429–442. [Google Scholar] [CrossRef]
- Lin, J.S.; Huang, X.X.; Li, Q.; Cao, Y.; Bao, Y.; Meng, X.F.; Li, Y.J.; Fu, C.; Hou, B.K. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016, 88, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.Z.; Jin, S.H.; Jiang, X.Y.; Dong, R.R.; Li, P.; Li, Y.J.; Hou, B.K. Ectopic expression of UGT75D1, a glycosyltransferase preferring indole-3-butyric acid, modulates cotyledon development and stress tolerance in seed germination of Arabidopsis thaliana. Plant. Mol. Biol. 2016, 90, 77–93. [Google Scholar] [CrossRef]
- Woo, H.H.; Jeong, B.R.; Hirsch, A.M.; Hawes, M.C. Characterization of Arabidopsis AtUGT85A and AtGUS gene families and their expression in rapidly dividing tissues. Genomics 2007, 90, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.M.; Venugopal, S.; Navarre, D.; Kachroo, A. Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol. 2011, 155, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Yu, D. Arabidopsis WRKY2 transcription factor mediates seed germination and postgermination arrest of development by abscisic acid. BMC Plant. Biol. 2009, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Kracher, B.; Ziegler, J.; Birkenbihl, R.P.; Somssich, I.E. Negative regulation of ABA signaling by WRKY33 is critical for Arabidopsis immunity towards Botrytis cinerea 2100. eLife 2015, 4, e07295. [Google Scholar] [CrossRef] [PubMed]
- Willmann, M.R.; Mehalick, A.J.; Packer, R.L.; Jenik, P.D. MicroRNAs regulate the timing of embryo maturation in Arabidopsis. Plant. Physiol. 2011, 155, 1871–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef]
- Panstruga, R.; Parker, J.E.; Schulze-Lefert, P. SnapShot, plant immune response pathways. Cell 2009, 136, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Robert-Seilaniantz, A.; Grant, M.; Jones, J.D. Hormone crosstalk in plant disease and defense: More than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 2011, 49, 317–343. [Google Scholar] [CrossRef]
- Zhang, K.L.; Liu, Q.S.; Kang, H.X.; Liu, X.M.; Chen, X.P.; Peng, Y.F.; Li, Y.H. Herbivore-induced rice resistance against rice blast mediated by salicylic acid. Insect Sci. 2020, 27, 49–57. [Google Scholar] [CrossRef]
- Künstler, A.; Kátay, G.; Gullner, G.; Király, L. Artificial elevation of glutathione contents in salicylic acid-deficient tobacco (Nicotiana tabacum cv. Xanthi NahG) reduces susceptibility to the powdery mildew pathogen Euoidium longipes. Plant. Biol. 2019, 22, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Wang, K.; Sun, L.; Xing, H.; Wang, S.; Li, L.; Chen, S.; Guo, H.S.; Zhang, J. The plant-specific transcription factors CBP60g and SARD1 are targeted by a verticillium secretory protein VDSCP41 to modulate immunity. eLife 2018, 7, e34902. [Google Scholar] [CrossRef]
- Xing, L.; Gao, L.; Chen, Q.; Pei, H.; Di, Z.; Xiao, J.; Wang, H.; Ma, L.; Chen, P.; Cao, A. Over-expressing a UDP-glucosyltransferase gene (Ta-UGT3) enhances Fusarium Head Blight resistance of wheat. Plant. Growth Regul. 2018, 84, 561–571. [Google Scholar] [CrossRef]
- Gatz, C. From pioneers to team players: TGA transcription factors provide a molecular link between different stress pathways. Mol. Plant. Microbe Interact. 2013, 26, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton, J.; Flors, V.; Mauch-Mani, B. The multifaceted role of ABA in disease resistance. Trends Plant. Sci. 2009, 14, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Mang, H.G.; Qian, W.; Zhu, Y.; Qian, J.; Kang, H.G.; Klessig, D.F.; Hua, J. Abscisic acid deficiency antagonizes high-temperature inhibition of disease resistance through enhancing nuclear accumulation of resistance proteins SNC1 and RPS4 in Arabidopsis. Plant. Cell 2012, 24, 1271–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Park, Y.J.; Seo, P.J.; Kim, J.H.; Sim, H.J.; Kim, S.G.; Park, C.M. Systemic immunity requires SnRK2.8-mediated nuclear import of NPR1 in arabidopsis. Plant Cell 2015, 27, 3425–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.R.; Huang, L.Y.; Zhang, F.; Zhu, L.H.; Zhou, Y.L.; Li, Z.K. Genome-wide phylogenetic analysis of stress-activated protein kinase genes in rice (OsSAPKs) and expression profiling in response to Xanthomonas oryzae pv. oryzicola Infection. Plant. Mol. Biol. Report. 2013, 31, 877–885. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; Tang, L.; Tong, X.; Wang, L.; Liu, L.; Huang, S.; Zhang, J. SAPK10-mediated phosphorylation on WRKY72 releases its suppression on jasmonic acid biosynthesis and bacterial blight resistance. iScience 2019, 16, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Akimoto-Tomiyama, C.; Tanabe, S.; Kajiwara, H.; Minami, E.; Ochiai, H. Loss of chloroplast-localized protein phosphatase 2Cs in Arabidopsis thaliana leads to enhancement of plant immunity and resistance to Xanthomonas campestris pv. campestris infection. Mol. Plant. Pathol. 2018, 19, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Piarulli, L.; Gadaleta, A.; Mangini, G.; Signorile, M.A.; Pasquini, M.; Blanco, A.; Simeone, R. Molecular identification of a new powdery mildew resistance gene on chromosome 2BS from Triticum turgidum ssp. dicoccum. Plant. Sci. 2012, 196, 101–106. [Google Scholar] [CrossRef]
- Agata, G.; Pasqualina, C.; Stefania, L.G.; Antonio, B.; Angelica, G. Map-based cloning of QFhb.mgb-2A identifies a WAK2 gene responsible for Fusarium Head Blight resistance in wheat. Sci. Rep. 2019, 9, 6929. [Google Scholar]
- Cui, X.Y.; Du, Y.T.; Fu, J.D.; Yu, T.F.; Wang, C.T.; Chen, M.; Chen, J.; Ma, Y.Z.; Xu, Z.S. Wheat CBL-interacting protein kinase 23 positively regulates drought stress and ABA responses. BMC Plant. Biol. 2018, 18, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; van Wersch, R.; Zhang, Y. Convergent and divergent signaling in PAMP-triggered immunity and effector-triggered immunity. Mol. Plant. Microbe Interact. 2018, 31, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.F.; Zhang, Z.W.; Yuan, S. Putative connections between nitrate reductase S-nitrosylation and NO synthesis under pathogen attacks and abiotic stresses. Front. Plant. Sci. 2018, 9, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olea, F.; Pérez-García, A.; Cantón, F.R.; Rivera, M.E.; Cañas, R.; Avila, C.; Cazorla, F.M.; Cánovas, F.M.; de Vicente, A. Up-regulation and localization of asparagine synthetase in tomato leaves infected by the bacterial pathogen Pseudomonas syringae. Plant. Cell Physiol. 2004, 45, 770–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Hong, Y.; Huang, L.; Liu, S.; Tian, L.; Dai, Y.; Cao, Z.; Huang, L.; Li, D.; Song, F. Virus-induced gene silencing-based functional analyses revealed the involvement of several putative trehalose-6-phosphate synthase/phosphatase genes in disease resistance against Botrytis cinerea and Pseudomonas syringae pv. tomato DC3000 in Tomato. Front. Plant. Sci. 2016, 47, 1176. [Google Scholar] [CrossRef]
- Henry, E.; Fung, N.; Liu, J.; Drakakaki, G.; Coaker, G. Beyond glycolysis: GAPDHs are multi-functional enzymes involved in regulation of ROS, autophagy, and plant immune responses. PLoS Genet. 2015, 11, e1005199. [Google Scholar] [CrossRef]
- Hua, J. Modulation of plant immunity by light, circadian rhythm, and temperature. Curr. Opin. Plant. Biol. 2013, 16, 406–413. [Google Scholar] [CrossRef]
- Voll, L.M.; Zell, M.B.; Engelsdorf, T.; Saur, A.; Wheeler, M.G.; Drincovich, M.F.; Weber, A.P.; Maurino, V.G. Loss of cytosolic NADP-malic enzyme 2 in Arabidopsis thaliana is associated with enhanced susceptibility to Colletotrichum higginsianum. New Phytol. 2012, 195, 189–202. [Google Scholar] [CrossRef]
- Furlan, G.; Nakagami, H.; Eschen-Lippold, L.; Jiang, X.; Majovsky, P.; Kowarschik, K.; Hoehenwarter, W.; Lee, J.; Trujillo, M. Changes in PUB22 ubiquitination modes triggered by MITOGEN-ACTIVATED PROTEIN KINASE3 dampen the immune response. Plant. Cell 2017, 29, 726–745. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, M.; Shirasu, K. Ubiquitination in plant immunity. Curr. Opin. Plant. Biol. 2010, 13, 402–408. [Google Scholar] [CrossRef]
- Fang, H.; Meng, Q.; Xu, J.; Tang, H.; Tang, S.; Zhang, H.; Huang, J. Knock-down of stress inducible OsSRFP1 encoding an E3 ubiquitin ligase with transcriptional activation activity confers abiotic stress tolerance through enhancing antioxidant protection in rice. Plant. Mol. Biol. 2015, 87, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Cao, Y.; Sun, X.; Espinoza, C.; Nguyen, C.T.; Liang, Y.; Stacey, G. Arabidopsis E3 ubiquitin ligase PLANT U-BOX13 (PUB13) regulates chitin receptor LYSIN MOTIF RECEPTOR KINASE5 (LYK5) protein abundance. New Phytol. 2017, 214, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Zentgraf, U. A HECT E3 ubiquitin ligase negatively regulates Arabidopsis leaf senescence through degradation of the transcription factor WRKY53. Plant J. 2010, 63, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, W.; Wang, J.; Wang, L.; Yao, W.; Yang, Y.; Xu, Y.; Ma, F.; Du, Y.; Wang, Y. The Chinese wild grapevine (Vitis pseudoreticulata) E3 ubiquitin ligase Erysiphe necator-induced RING finger protein 1 (EIRP1) activates plant defense responses by inducing proteolysis of the VpWRKY11 transcription factor. New Phytol. 2013, 200, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2011, 25, 402–408. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Annotation | Triticum Aestivum ID (Chinese Spring) | Ortholog ID (Arabidopsis) | Function | |
|---|---|---|---|---|
| protein phosphorylation | receptor-like protein kinase 5 | TraesCS1D02G344700.1 | AT5G25930.1 | membrane signal transduction [20] |
| serine/threonine- kinase receptor | TraesCS5A02G189200.1 | AT3G15610.1 | plant developmental [21] | |
| hexokinase | TraesCS3D02G276200.1 | AT5G25930.1 | membrane signal transduction [20] | |
| interleukin-1 receptor-associated kinase | TraesCS4D02G286100.1 | AT2G02800.1 | early elicitor signaling [22] | |
| serine/threonine-protein kinase | TraesCS7D02G165300.1 | AT2G05940.1 | PTI [23] | |
| serine/threonine-protein kinase SD1-29 | TraesCS2D02G217400.1 | AT4G21390.2 | early elicitor signaling [22] | |
| protein kinase APK1A | TraesCS7A02G163300.1 | AT2G05940.1 | PTI [23] | |
| serine/threonine-protein kinase PBS1 | TraesCS2A02G348300.1 | AT3G59360.1 | cell wall biosynthesis [24] | |
| receptor-like protein kinase FERONIA | TraesCS5D02G336300.1 | AT3G51550.1 | male–female interaction [25] | |
| inactive receptor kinase | TraesCS4A02G267300.1 | AT1G48480.1 | pathogen infection [26] | |
| flavonoid biosynthetic | salicylic acid glucosyltransferase | TraesCS5A02G149600.1 | AT1G05675.1 | SA/JA-mediated defense [27] |
| hydroquinone glucosyltransferase-like | TraesCS7A02G216000.1 | AT4G01070.1 | cell wall lignification [28] | |
| crocetin glucosyltransferase | TraesCS3D02G120200.1 | AT4G15550.1 | seed germination [29] | |
| cyanohydrin beta-glucosyltransferase | TraesCS5D02G324600.1 | AT1G22360.1 | cell cycle regulation [30] | |
| anthocyanidin 5, 3-O-glucosyltransferase | TraesCS2B02G012000.1 | AT3G16520.3 | unknown | |
| protein O-GlcNAc transferase | TraesCS1A02G351900.1 | AT3G18170.1 | unknown | |
| protein O-GlcNAc transferase | TraesCS1D02G358500.1 | AT3G18170.1 | unknown | |
| signaling transcription | WRKY transcription factor 2 | TraesCS3A02G209800.1 | AT5G26170.1 | JA defense responses [31] |
| WRKY transcription factor 2 | TraesCS5B02G257300.1 | AT5G56270.1 | seed germination by ABA [32] | |
| WRKY transcription factor 33 | TraesCS1D02G292700.1 | AT2G38470.1 | ABA signaling [33] | |
| oxidative stress | peroxidase 55 precursor | TraesCS5B02G287600.1 | AT5G14130.1 | unknown |
| peroxidase | TraesCS7B02G381400.1 | AT4G39720.1 | unknown | |
| peroxidase | TraesCS1B02G420500.1 | AT3G14180.1 | seedling development [34] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Sun, L.; Chen, Y.; Wei, L.; Hao, Y.; Yu, Z.; Wang, Z.; Zhang, H.; Zhang, X.; Li, M.; et al. The Regulatory Network of CMPG1-V in Wheat–Blumeria graminis f. sp. tritici Interaction Revealed by Temporal Profiling Using RNA-Seq. Int. J. Mol. Sci. 2020, 21, 5967. https://doi.org/10.3390/ijms21175967
Liu J, Sun L, Chen Y, Wei L, Hao Y, Yu Z, Wang Z, Zhang H, Zhang X, Li M, et al. The Regulatory Network of CMPG1-V in Wheat–Blumeria graminis f. sp. tritici Interaction Revealed by Temporal Profiling Using RNA-Seq. International Journal of Molecular Sciences. 2020; 21(17):5967. https://doi.org/10.3390/ijms21175967
Chicago/Turabian StyleLiu, Jia, Li Sun, Yiming Chen, Luyang Wei, Yongli Hao, Zhongyu Yu, Zongkuan Wang, Heng Zhang, Xu Zhang, Mengli Li, and et al. 2020. "The Regulatory Network of CMPG1-V in Wheat–Blumeria graminis f. sp. tritici Interaction Revealed by Temporal Profiling Using RNA-Seq" International Journal of Molecular Sciences 21, no. 17: 5967. https://doi.org/10.3390/ijms21175967
APA StyleLiu, J., Sun, L., Chen, Y., Wei, L., Hao, Y., Yu, Z., Wang, Z., Zhang, H., Zhang, X., Li, M., Wang, H., Xiao, J., & Wang, X. (2020). The Regulatory Network of CMPG1-V in Wheat–Blumeria graminis f. sp. tritici Interaction Revealed by Temporal Profiling Using RNA-Seq. International Journal of Molecular Sciences, 21(17), 5967. https://doi.org/10.3390/ijms21175967