
The Genetic Landscape of Parkinsonism-Related Dystonias and Atypical Parkinsonism-Related Syndromes

Abstract
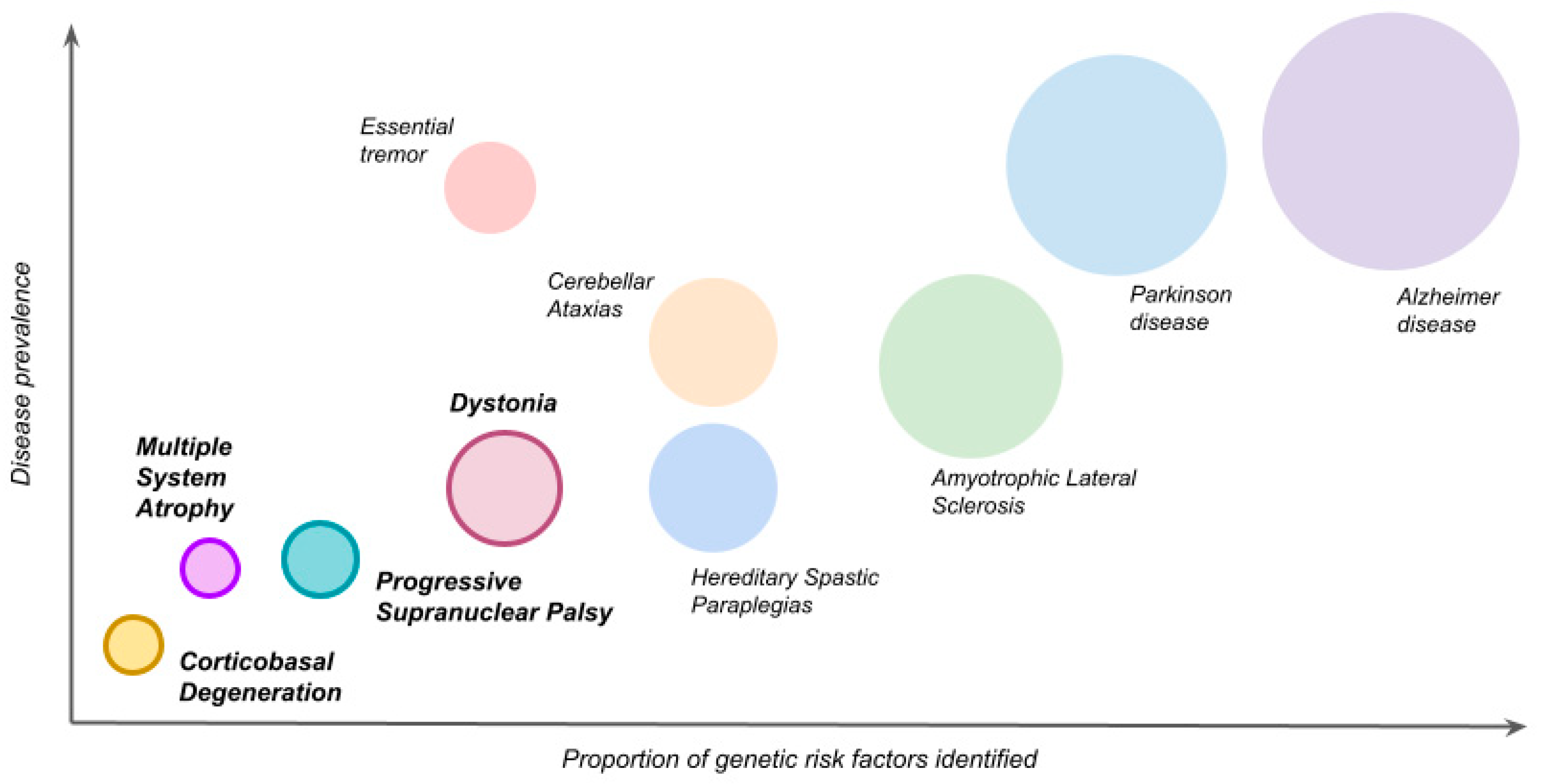
:1. Introduction
2. Parkinsonism-Related Movement Disorders
Dystonia-Plus Syndromes
3. Atypical Parkinsonism-Related Neurodegenerative Disorders
3.1. Progressive Supranuclear Palsy
3.2. Multiple System Atrophy
3.3. Corticobasal Degeneration
4. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Epidemiological Study of Dystonia in Europe (ESDE). Collaborative Group A Prevalence Study of Primary Dystonia in Eight European Countries. J. Neurol. 2000, 247, 787–792. [Google Scholar] [CrossRef]
- Müller, J.; Kiechl, S.; Wenning, G.K.; Seppi, K.; Willeit, J.; Gasperi, A.; Wissel, J.; Gasser, T.; Poewe, W. The Prevalence of Primary Dystonia in the General Community. Neurology 2002, 59, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Grütz, K.; Klein, C. Dystonia Updates: Definition, Nomenclature, Clinical Classification, and Etiology. J. Neural Transm. 2021, 128, 395–404. [Google Scholar] [CrossRef]
- Albanese, A.; Bhatia, K.; Bressman, S.B.; Delong, M.R.; Fahn, S.; Fung, V.S.C.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and Classification of Dystonia: A Consensus Update. Mov. Disord. 2013, 28, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Shetty, A.S.; Bhatia, K.P.; Lang, A.E. Dystonia and Parkinson’s Disease: What Is the Relationship? Neurobiol. Dis. 2019, 132, 104462. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Mulroy, E.; Gövert, F.; Latorre, A.; Di Lazarro, G.; Erro, R.; Batla, A.; Holton, J.L.; Miki, Y.; Warner, T.T.; et al. Development of Parkinsonism after Long-Standing Cervical Dystonia-A Cohort. J. Neurol. Sci. 2021, 427, 117477. [Google Scholar] [CrossRef]
- Kidron, D.; Melamed, E. Forms of Dystonia in Patients with Parkinson’s Disease. Neurology 1987, 37, 1009–1011. [Google Scholar] [CrossRef]
- Grünewald, A.; Kasten, M.; Ziegler, A.; Klein, C. Next-Generation Phenotyping Using the Parkin Example: Time to Catch up with Genetics. JAMA Neurol. 2013, 70, 1186–1191. [Google Scholar] [CrossRef]
- Elia, A.E.; Del Sorbo, F.; Romito, L.M.; Barzaghi, C.; Garavaglia, B.; Albanese, A. Isolated Limb Dystonia as Presenting Feature of Parkin Disease. J. Neurol. Neurosurg. Psychiatry 2014, 85, 827–828. [Google Scholar] [CrossRef]
- Poewe, W.H.; Lees, A.J.; Stern, G.M. Dystonia in Parkinson’s Disease: Clinical and Pharmacological Features. Ann. Neurol. 1988, 23, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Phukan, J.; Albanese, A.; Gasser, T.; Warner, T. Primary Dystonia and Dystonia-plus Syndromes: Clinical Characteristics, Diagnosis, and Pathogenesis. Lancet Neurol. 2011, 10, 1074–1085. [Google Scholar] [CrossRef]
- Thongchuam, Y.; Panyakaew, P.; Bhidayasiri, R. Orofacial Dystonia and Asssociated Bulbar Symptoms in Multiple System Atrophy: A Blinded Video Analysis. J. Neurol. Sci. 2020, 417, 116992. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.T. Comparison of Dystonia between Parkinson’s Disease and Atypical Parkinsonism: The Clinical Usefulness of Dystonia Distribution and Characteristics in the Differential Diagnosis of Parkinsonism. Neurol. Neurochir. Polska 2018, 52, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, L.M.; Junker, J.; Loens, S.; Baumann, H.; Olschewski, L.; Schaake, S.; Madoev, H.; Petkovic, S.; Kuhnke, N.; Kasten, M.; et al. Genotype-Phenotype Relations for Isolated Dystonia Genes: MDSGene Systematic Review. Mov. Disord. 2021, 36, 1086–1103. [Google Scholar] [CrossRef] [PubMed]
- Zech, M.; Jech, R.; Boesch, S.; Škorvánek, M.; Weber, S.; Wagner, M.; Zhao, C.; Jochim, A.; Necpál, J.; Dincer, Y.; et al. Monogenic Variants in Dystonia: An Exome-Wide Sequencing Study. Lancet Neurol. 2020, 19, 908–918. [Google Scholar] [CrossRef]
- Zech, M.; Boesch, S.; Škorvánek, M.; Necpál, J.; Švantnerová, J.; Wagner, M.; Dincer, Y.; Sadr-Nabavi, A.; Serranová, T.; Rektorová, I.; et al. Clinically Relevant Copy-Number Variants in Exome Sequencing Data of Patients with Dystonia. Parkinsonism Relat. Disord. 2021, 84, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.H.; Gibb, W.R.; Zhong, X.H.; Shannak, K.S.; Kish, S.; Chang, L.G.; Hornykiewicz, O. Dopa-Responsive Dystonia: Pathological and Biochemical Observations in a Case. Ann. Neurol. 1994, 35, 396–402. [Google Scholar] [CrossRef]
- Ichinose, H.; Ohye, T.; Takahashi, E.; Seki, N.; Hori, T.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S. Hereditary Progressive Dystonia with Marked Diurnal Fluctuation Caused by Mutations in the GTP Cyclohydrolase I Gene. Nat. Genet. 1994, 8, 236–242. [Google Scholar] [CrossRef]
- Mencacci, N.E.; Isaias, I.U.; Reich, M.M.; Ganos, C.; Plagnol, V.; Polke, J.M.; Bras, J.; Hersheson, J.; Stamelou, M.; Pittman, A.M.; et al. Parkinson’s Disease in GTP Cyclohydrolase 1 Mutation Carriers. Brain 2014, 137, 2480–2492. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Rudakou, U.; Bencheikh, B.O.A.; Ruskey, J.A.; Krohn, L.; Laurent, S.B.; Spiegelman, D.; Liong, C.; Fahn, S.; Waters, C.; Monchi, O.; et al. Common and Rare GCH1 Variants Are Associated with Parkinson’s Disease. Neurobiol. Aging 2019, 73, 231.e1–231.e6. [Google Scholar] [CrossRef]
- Tadic, V.; Kasten, M.; Brüggemann, N.; Stiller, S.; Hagenah, J.; Klein, C. Dopa-Responsive Dystonia Revisited: Diagnostic Delay, Residual Signs, and Nonmotor Signs. Arch. Neurol. 2012, 69, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.A.; Barclay, C.L.; Duff, J.; Furukawa, Y.; Lang, A.E. Phenocopies in a Large GCH1 Mutation Positive Family with Dopa Responsive Dystonia: Confusing the Picture? J. Neurol. Neurosurg. Psychiatry 2002, 72, 801–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, S.; Naiya, T.; Roy, S.; Das, G.; Wali, G.M.; Das, S.K.; Ray, K.; Ray, J. A Compound Heterozygote for GCH1 Mutation Represents a Case of Atypical Dopa-Responsive Dystonia. J. Mol. Neurosci. 2019, 68, 214–220. [Google Scholar] [CrossRef]
- Lüdecke, B.; Dworniczak, B.; Bartholomé, K. A Point Mutation in the Tyrosine Hydroxylase Gene Associated with Segawa’s Syndrome. Hum. Genet. 1995, 95, 123–125. [Google Scholar] [CrossRef]
- Cai, C.; Shi, W.; Zeng, Z.; Zhang, M.; Ling, C.; Chen, L.; Cai, C.; Zhang, B.; Li, W.-D. GTP Cyclohydrolase I and Tyrosine Hydroxylase Gene Mutations in Familial and Sporadic Dopa-Responsive Dystonia Patients. PLoS ONE 2013, 8, e65215. [Google Scholar] [CrossRef] [PubMed]
- Rodacker, V.; Toustrup-Jensen, M.; Vilsen, B. Mutations Phe785Leu and Thr618Met in Na+,K+-ATPase, Associated with Familial Rapid-Onset Dystonia Parkinsonism, Interfere with Na+ Interaction by Distinct Mechanisms. J. Biol. Chem. 2006, 281, 18539–18548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon, D.P.; Fremont, R.; Kraenzlin, F.; Khodakhah, K. The Neural Substrates of Rapid-Onset Dystonia-Parkinsonism. Nat. Neurosci. 2011, 14, 357–365. [Google Scholar] [CrossRef]
- Camargos, S.; Scholz, S.; Simón-Sánchez, J.; Paisán-Ruiz, C.; Lewis, P.; Hernandez, D.; Ding, J.; Gibbs, J.R.; Cookson, M.R.; Bras, J.; et al. DYT16, a Novel Young-Onset Dystonia-Parkinsonism Disorder: Identification of a Segregating Mutation in the Stress-Response Protein PRKRA. Lancet Neurol. 2008, 7, 207–215. [Google Scholar] [CrossRef]
- Seibler, P.; Djarmati, A.; Langpap, B.; Hagenah, J.; Schmidt, A.; Brüggemann, N.; Siebner, H.; Jabusch, H.-C.; Altenmüller, E.; Münchau, A.; et al. A Heterozygous Frameshift Mutation in PRKRA (DYT16) Associated with Generalised Dystonia in a German Patient. Lancet Neurol. 2008, 7, 380–381. [Google Scholar] [CrossRef]
- Zech, M.; Castrop, F.; Schormair, B.; Jochim, A.; Wieland, T.; Gross, N.; Lichtner, P.; Peters, A.; Gieger, C.; Meitinger, T.; et al. DYT16 Revisited: Exome Sequencing Identifies PRKRA Mutations in a European Dystonia Family. Mov. Disord. 2014, 29, 1504–1510. [Google Scholar] [CrossRef]
- Quadri, M.; Olgiati, S.; Sensi, M.; Gualandi, F.; Groppo, E.; Rispoli, V.; Graafland, J.; Breedveld, G.J.; Fabbrini, G.; Berardelli, A.; et al. PRKRA Mutation Causing Early-Onset Generalized Dystonia-Parkinsonism (DYT16) in an Italian Family. Mov. Disord. 2016, 31, 765–767. [Google Scholar] [CrossRef]
- Dos Santos, C.O.; da Silva-Júnior, F.P.; Puga, R.D.; Barbosa, E.R.; Silva, S.M.C.A.; Borges, V.; Limongi, J.C.P.; Rocha, M.S.G.; Ferraz, H.B.; de Carvalho Aguiar, P. The Prevalence of PRKRA Mutations in Idiopathic Dystonia. Parkinsonism Relat. Disord. 2018, 48, 93–96. [Google Scholar] [CrossRef]
- Kurian, M.A.; Li, Y.; Zhen, J.; Meyer, E.; Hai, N.; Christen, H.-J.; Hoffmann, G.F.; Jardine, P.; von Moers, A.; Mordekar, S.R.; et al. Clinical and Molecular Characterisation of Hereditary Dopamine Transporter Deficiency Syndrome: An Observational Cohort and Experimental Study. Lancet Neurol. 2011, 10, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Kaji, R.; Ando, S.; Tomizawa, M.; Yasuno, K.; Goto, S.; Matsumoto, S.; Tabuena, M.D.; Maranon, E.; Dantes, M.; et al. Reduced Neuron-Specific Expression of the TAF1 Gene Is Associated with X-Linked Dystonia-Parkinsonism. Am. J. Hum. Genet. 2007, 80, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragg, D.C.; Mangkalaphiban, K.; Vaine, C.A.; Kulkarni, N.J.; Shin, D.; Yadav, R.; Dhakal, J.; Ton, M.-L.; Cheng, A.; Russo, C.T.; et al. Disease Onset in X-Linked Dystonia-Parkinsonism Correlates with Expansion of a Hexameric Repeat within an SVA Retrotransposon in. Proc. Natl. Acad. Sci. USA 2017, 114, E11020–E11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westenberger, A.; Reyes, C.J.; Saranza, G.; Dobricic, V.; Hanssen, H.; Domingo, A.; Laabs, B.-H.; Schaake, S.; Pozojevic, J.; Rakovic, A.; et al. A Hexanucleotide Repeat Modifies Expressivity of X-Linked Dystonia Parkinsonism. Ann. Neurol. 2019, 85, 812–822. [Google Scholar] [CrossRef]
- Laabs, B.-H.; Klein, C.; Pozojevic, J.; Domingo, A.; Brüggemann, N.; Grütz, K.; Rosales, R.L.; Jamora, R.D.; Saranza, G.; Diesta, C.C.E.; et al. Identifying Genetic Modifiers of Age-Associated Penetrance in X-Linked Dystonia-Parkinsonism. Nat. Commun. 2021, 12, 3216. [Google Scholar] [CrossRef]
- Lalli, S.; Albanese, A. The Diagnostic Challenge of Primary Dystonia: Evidence from Misdiagnosis. Mov. Disord. 2010, 25, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Klepitskaya, O.; Neuwelt, A.J.; Nguyen, T.; Leehey, M. Primary Dystonia Misinterpreted as Parkinson Disease: Video Case Presentation and Practical Clues. Neurol. Clin. Pract. 2013, 3, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Luciano, M.S.; Ozelius, L.; Sims, K.; Raymond, D.; Liu, L.; Saunders-Pullman, R. Responsiveness to Levodopa in Epsilon-Sarcoglycan Deletions. Mov. Disord. 2009, 24, 425–428. [Google Scholar] [CrossRef]
- Armstrong, M.J. Progressive Supranuclear Palsy: An Update. Curr. Neurol. Neurosci. Rep. 2018, 18, 12. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Chambers, C.B.; Lee, J.M.; Troncoso, J.C.; Reich, S.; Muma, N.A. Overexpression of Four-Repeat Tau mRNA Isoforms in Progressive Supranuclear Palsy but Not in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 325–332. [Google Scholar] [CrossRef]
- Ling, H. Clinical Approach to Progressive Supranuclear Palsy. J. Mov. Disord. 2016, 9, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Takigawa, H.; Kitayama, M.; Wada-Isoe, K.; Kowa, H.; Nakashima, K. Prevalence of Progressive Supranuclear Palsy in Yonago: Change throughout a Decade. Brain Behav. 2016, 6, e00557. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Melhem, N.M.; Dickson, D.W.; Sleiman, P.M.A.; Wang, L.-S.; Klei, L.; Rademakers, R.; de Silva, R.; Litvan, I.; Riley, D.E.; et al. Identification of Common Variants Influencing Risk of the Tauopathy Progressive Supranuclear Palsy. Nat. Genet. 2011, 43, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.A.; Chen, Z.; Won, H.; Huang, A.Y.; Lowe, J.K.; Wojta, K.; Yokoyama, J.S.; Bensimon, G.; Leigh, P.N.; Payan, C.; et al. Joint Genome-Wide Association Study of Progressive Supranuclear Palsy Identifies Novel Susceptibility Loci and Genetic Correlation to Neurodegenerative Diseases. Mol. Neurodegener. 2018, 13, 41. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.A.; Shatunov, A.; Jones, A.R.; Kravitz, S.N.; Huang, A.Y.; Lawrence, L.; Lowe, J.K.; Lewis, C.M.; Payan, C.A.M.; et al. Genome-Wide Survey of Copy Number Variants Finds MAPT Duplications in Progressive Supranuclear Palsy. Mov. Disord. 2019, 34, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, J.D.; Paviour, D.; Vandrovcova, J.; Hodges, J.; de Silva, R.; Rossor, M.N. Novel L284R MAPT Mutation in a Family with an Autosomal Dominant Progressive Supranuclear Palsy Syndrome. Neurodegener. Dis. 2011, 8, 149–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, R.; Thobois, S.; Streichenberger, N.; Kopp, N.; Sánchez, M.P.; Pérez, M.; Hoenicka, J.; Avila, J.; Honnorat, J.; de Yébenes, J.G. A New Mutation of the Tau Gene, G303V, in Early-Onset Familial Progressive Supranuclear Palsy. Arch. Neurol. 2005, 62, 1444–1450. [Google Scholar] [CrossRef]
- Forrest, S.L.; Halliday, G.M.; McCann, H.; McGeachie, A.B.; McGinley, C.V.; Hodges, J.R.; Piguet, O.; Kwok, J.B.; Spillantini, M.G.; Kril, J.J. Heritability in Frontotemporal Tauopathies. Alzheimers Dement. 2019, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kaat, L.D.; Boon, A.J.W.; Azmani, A.; Kamphorst, W.; Breteler, M.M.B.; Anar, B.; Heutink, P.; van Swieten, J.C. Familial Aggregation of Parkinsonism in Progressive Supranuclear Palsy. Neurology 2009, 73, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical Diagnosis of Progressive Supranuclear Palsy: The Movement Disorder Society Criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhou, Y.; Jiao, B.; Shen, L. Genetics of Progressive Supranuclear Palsy: A Review. J. Parkinsons Dis. 2021, 11, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Kawamata, T.; Komure, O.; Kuno, S.; D’Souza, I.; Poorkaj, P.; Kawai, J.; Tanimukai, S.; Yamamoto, Y.; Hasegawa, H.; et al. A Mutation in the Microtubule-Associated Protein Tau in Pallido-Nigro-Luysian Degeneration. Neurology 1999, 53, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Soliveri, P.; Rossi, G.; Monza, D.; Tagliavini, F.; Piacentini, S.; Albanese, A.; Bugiani, O.; Girotti, F. A Case of Dementia Parkinsonism Resembling Progressive Supranuclear Palsy due to Mutation in the Tau Protein Gene. Arch. Neurol. 2003, 60, 1454–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogaki, K.; Li, Y.; Takanashi, M.; Ishikawa, K.-I.; Kobayashi, T.; Nonaka, T.; Hasegawa, M.; Kishi, M.; Yoshino, H.; Funayama, M.; et al. Analyses of the MAPT, PGRN, and C9orf72 Mutations in Japanese Patients with FTLD, PSP, and CBS. Parkinsonism Relat. Disord. 2013, 19, 15–20. [Google Scholar] [CrossRef]
- Nakayama, S.; Shimonaka, S.; Elahi, M.; Nishioka, K.; Oji, Y.; Matsumoto, S.-E.; Li, Y.; Yoshino, H.; Mogushi, K.; Hatano, T.; et al. Tau Aggregation and Seeding Analyses of Two Novel MAPT Variants Found in Patients with Motor Neuron Disease and Progressive Parkinsonism. Neurobiol. Aging 2019, 84, 240.e13–240.e22. [Google Scholar] [CrossRef]
- Fujioka, S.; Sanchez Contreras, M.Y.; Strongosky, A.J.; Ogaki, K.; Whaley, N.R.; Tacik, P.M.; van Gerpen, J.A.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K.; et al. Three Sib-Pairs of Autopsy-Confirmed Progressive Supranuclear Palsy. Parkinsonism Relat. Disord. 2015, 21, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, A.; Ash, E.; Gan-Or, Z.; Orr-Urtreger, A.; Giladi, N. The LRRK2 G2019S Mutation as the Cause of Parkinson’s Disease in Ashkenazi Jews. J. Neural Transm. 2009, 116, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, E.; Koga, S.; Valentino, R.R.; Reynolds, R.H.; Ferrari, R.; Tan, M.M.X.; Rowe, J.B.; Dalgard, C.L.; Scholz, S.W.; Dickson, D.W.; et al. Genetic Determinants of Survival in Progressive Supranuclear Palsy: A Genome-Wide Association Study. Lancet Neurol. 2021, 20, 107–116. [Google Scholar] [CrossRef]
- Rajput, A.; Dickson, D.W.; Robinson, C.A.; Ross, O.A.; Dächsel, J.C.; Lincoln, S.J.; Cobb, S.A.; Rajput, M.L.; Farrer, M.J. Parkinsonism, Lrrk2 G2019S, and Tau Neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef]
- Ruffmann, C.; Giaccone, G.; Canesi, M.; Bramerio, M.; Goldwurm, S.; Gambacorta, M.; Rossi, G.; Tagliavini, F.; Pezzoli, G. Atypical Tauopathy in a Patient with LRRK2-G2019S Mutation and Tremor-Dominant Parkinsonism. Neuropathol. Appl. Neurobiol. 2012, 38, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Heckman, M.G.; Tacik, P.; Diehl, N.; Brown, P.H.; Soto-Ortolaza, A.I.; Christopher, E.A.; Walton, R.L.; Ross, O.A.; Golbe, L.I.; et al. Study of LRRK2 Variation in Tauopathy: Progressive Supranuclear Palsy and Corticobasal Degeneration. Mov. Disord. 2017, 32, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Pletnikova, O.; Geiger, J.T.; Murphy, N.A.; Abramzon, Y.; Rudow, G.; Mamais, A.; Sabir, M.S.; Crain, B.; Ahmed, S.; et al. Genetic Analysis of Neurodegenerative Diseases in a Pathology Cohort. Neurobiol. Aging 2019, 76, 214.e1–214.e9. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Wszolek, Z.K.; Pfeiffer, R.F.; Tsuboi, Y.; Uitti, R.J.; McComb, R.D.; Stoessl, A.J.; Strongosky, A.J.; Zimprich, A.; Müller-Myhsok, B.; Farrer, M.J.; et al. Autosomal Dominant Parkinsonism Associated with Variable Synuclein and Tau Pathology. Neurology 2004, 62, 1619–1622. [Google Scholar] [CrossRef]
- Spanaki, C.; Latsoudis, H.; Plaitakis, A. LRRK2 Mutations on Crete: R1441H Associated with PD Evolving to PSP. Neurology 2006, 67, 1518–1519. [Google Scholar] [CrossRef]
- Trinh, J.; Guella, I.; McKenzie, M.; Gustavsson, E.K.; Szu-Tu, C.; Petersen, M.S.; Rajput, A.; Rajput, A.H.; McKeown, M.; Jeon, B.S.; et al. Novel LRRK2 Mutations in Parkinsonism. Parkinsonism Relat. Disord. 2015, 21, 1119–1121. [Google Scholar] [CrossRef]
- Caroppo, P.; Le Ber, I.; Clot, F.; Rivaud-Péchoux, S.; Camuzat, A.; De Septenville, A.; Boutoleau-Bretonnière, C.; Mourlon, V.; Sauvée, M.; Lebouvier, T.; et al. DCTN1 Mutation Analysis in Families with Progressive Supranuclear Palsy-like Phenotypes. JAMA Neurol. 2014, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, E.K.; Trinh, J.; Guella, I.; Szu-Tu, C.; Khinda, J.; Lin, C.-H.; Wu, R.-M.; Stoessl, J.; Appel-Cresswell, S.; McKeown, M.; et al. DCTN1 p.K56R in Progressive Supranuclear Palsy. Parkinsonism Relat. Disord. 2016, 28, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Sasagasako, N.; Shen, C.; Shijo, M.; Hamasaki, H.; Suzuki, S.O.; Tsuboi, Y.; Fujii, N.; Iwaki, T. DCTN1 F52L Mutation Case of Perry Syndrome with Progressive Supranuclear Palsy-like Tauopathy. Parkinsonism Relat. Disord. 2018, 51, 105–110. [Google Scholar] [CrossRef]
- Yabe, I.; Yaguchi, H.; Kato, Y.; Miki, Y.; Takahashi, H.; Tanikawa, S.; Shirai, S.; Takahashi, I.; Kimura, M.; Hama, Y.; et al. Mutations in Bassoon in Individuals with Familial and Sporadic Progressive Supranuclear Palsy-like Syndrome. Sci. Rep. 2018, 8, 819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, B.; Martínez, A.; Gonzalo, I.; Vidal, L.; Ros, R.; Gomez-Tortosa, E.; Rabano, A.; Ampuero, I.; Sánchez, M.; Hoenicka, J.; et al. Steele-Richardson-Olszewski Syndrome in a Patient with a Single C212Y Mutation in the Parkin Protein. Mov. Disord. 2002, 17, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.P.; Gonzalo, I.; Avila, J.; De Yébenes, J.G. Progressive Supranuclear Palsy and Tau Hyperphosphorylation in a Patient with a C212Y Parkin Mutation. J. Alzheimers Dis. 2002, 4, 399–404. [Google Scholar] [CrossRef]
- Lesage, S.; Le Ber, I.; Condroyer, C.; Broussolle, E.; Gabelle, A.; Thobois, S.; Pasquier, F.; Mondon, K.; Dion, P.A.; Rochefort, D.; et al. C9orf72 Repeat Expansions Are a Rare Genetic Cause of Parkinsonism. Brain 2013, 136, 385–391. [Google Scholar] [CrossRef]
- Le Ber, I.; Camuzat, A.; Guillot-Noel, L.; Hannequin, D.; Lacomblez, L.; Golfier, V.; Puel, M.; Martinaud, O.; Deramecourt, V.; Rivaud-Pechoux, S.; et al. C9ORF72 Repeat Expansions in the Frontotemporal Dementias Spectrum of Diseases: A Flow-Chart for Genetic Testing. J. Alzheimers. Dis. 2013, 34, 485–499. [Google Scholar] [CrossRef] [Green Version]
- Wilke, C.; Pomper, J.K.; Biskup, S.; Puskás, C.; Berg, D.; Synofzik, M. Atypical Parkinsonism in C9orf72 Expansions: A Case Report and Systematic Review of 45 Cases from the Literature. J. Neurol. 2016, 263, 558–574. [Google Scholar] [CrossRef]
- Yabe, I.; Nakano, F.; Shirai, S.; Matsushima, M.; Takahashi, I.; Sasaki, H. Frontotemporal Dementia and Progressive Supranuclear Palsy-like Syndrome with a novelTARDBPmutation. Neurol. Clin. Neurosci. 2016, 4, 76–77. [Google Scholar] [CrossRef] [Green Version]
- Godeiro-Júnior, C.; Inaoka, R.J.; Barbosa, M.R.; Silva, M.R.R.; Aguiar, P.D.C.; Barsottini, O. Mutations in NPC1 in Two Brazilian Patients with Niemann-Pick Disease Type C and Progressive Supranuclear Palsy-like Presentation. Mov. Disord. 2006, 21, 2270–2272. [Google Scholar] [CrossRef]
- Cupidi, C.; Frangipane, F.; Gallo, M.; Clodomiro, A.; Colao, R.; Bernardi, L.; Anfossi, M.; Conidi, M.E.; Vasso, F.; Curcio, S.A.M.; et al. Role of Niemann-Pick Type C Disease Mutations in Dementia. J. Alzheimers Dis. 2017, 55, 1249–1259. [Google Scholar] [CrossRef]
- Tremolizzo, L.; Bertola, F.; Casati, G.; Piperno, A.; Ferrarese, C.; Appollonio, I. Progressive Supranuclear Palsy-like Phenotype Caused by Progranulin p.Thr272fs Mutation. Mov. Disord. 2011, 26, 1964–1966. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.; Baets, J.; De Bleecker, J.L.; Deconinck, T.; Biskup, S.; Hayer, S.N.; Züchner, S.; Schüle, R.; De Jonghe, P.; Synofzik, M. Beyond ALS and FTD: The Phenotypic Spectrum of TBK1 Mutations Includes PSP-like and Cerebellar Phenotypes. Neurobiol. Aging 2018, 62, 244.e9–244.e13. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, E.; Woodside, J.; Tan, M.M.X.; Shoai, M.; Pittman, A.; Ferrari, R.; Mok, K.Y.; Zhang, D.; Reynolds, R.H.; de Silva, R.; et al. Variation at the TRIM11 Locus Modifies Progressive Supranuclear Palsy Phenotype. Ann. Neurol. 2018, 84, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Katzeff, J.S.; Phan, K.; Purushothuman, S.; Halliday, G.M.; Kim, W.S. Cross-Examining Candidate Genes Implicated in Multiple System Atrophy. Acta Neuropathol. Commun. 2019, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A.; Wenning, G.K. Multiple System Atrophy: Pathogenic Mechanisms and Biomarkers. J. Neural Transm. 2016, 123, 555–572. [Google Scholar] [CrossRef]
- Federoff, M.; Price, T.R.; Sailer, A.; Scholz, S.; Hernandez, D.; Nicolas, A.; Singleton, A.B.; Nalls, M.; Houlden, H. Genome-Wide Estimate of the Heritability of Multiple System Atrophy. Parkinsonism Relat. Disord. 2016, 22, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemberger, S.; Scholz, S.W.; Singleton, A.B.; Wenning, G.K. Genetic Players in Multiple System Atrophy: Unfolding the Nature of the Beast. Neurobiol. Aging 2011, 32, 1924.e5–1924.e14. [Google Scholar] [CrossRef] [Green Version]
- Scholz, S.W.; Houlden, H.; Schulte, C.; Sharma, M.; Li, A.; Berg, D.; Melchers, A.; Paudel, R.; Gibbs, J.R.; Simon-Sanchez, J.; et al. SNCA Variants Are Associated with Increased Risk for Multiple System Atrophy. Ann. Neurol. 2009, 65, 610–614. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Dürr, A.; Wood, N.W.; Parkinson, M.H.; Camuzat, A.; Hulot, J.-S.; Morrison, K.E.; Renton, A.; Sussmuth, S.D.; Landwehrmeyer, B.G.; et al. Genetic Variants of the Alpha-Synuclein Gene SNCA Are Associated with Multiple System Atrophy. PLoS ONE 2009, 4, e7114. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wei, Q.-Q.; Ou, R.; Cao, B.; Chen, X.; Zhao, B.; Guo, X.; Yang, Y.; Chen, K.; Wu, Y.; et al. Genetic Variants of SNCA Are Associated with Susceptibility to Parkinson’s Disease but Not Amyotrophic Lateral Sclerosis or Multiple System Atrophy in a Chinese Population. PLoS ONE 2015, 10, e0133776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multiple-System Atrophy Research Collaboration Mutations in COQ2 in Familial and Sporadic Multiple-System Atrophy. N. Engl. J. Med. 2013, 369, 233–244. [CrossRef]
- Zhao, Q.; Yang, X.; Tian, S.; An, R.; Zheng, J.; Xu, Y. Association of the COQ2 V393A Variant with Risk of Multiple System Atrophy in East Asians: A Case-Control Study and Meta-Analysis of the Literature. Neurol. Sci. 2016, 37, 423–430. [Google Scholar] [CrossRef]
- Sailer, A.; Scholz, S.W.; Nalls, M.A.; Schulte, C.; Federoff, M.; Price, T.R.; Lees, A.; Ross, O.A.; Dickson, D.W.; Mok, K.; et al. A Genome-Wide Association Study in Multiple System Atrophy. Neurology 2016, 87, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, J.; Matsukawa, T.; Sasaki, H.; Yabe, I.; Matsushima, M.; Dürr, A.; Brice, A.; Takashima, H.; Kikuchi, A.; Aoki, M.; et al. Variants Associated with Gaucher Disease in Multiple System Atrophy. Ann. Clin. Transl. Neurol. 2015, 2, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sklerov, M.; Kang, U.J.; Liong, C.; Clark, L.; Marder, K.; Pauciulo, M.; Nichols, W.C.; Chung, W.K.; Honig, L.S.; Cortes, E.; et al. Frequency of Variants in Autopsy-Proven Multiple System Atrophy. Mov. Disord. Clin. Pract. 2017, 4, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Segarane, B.; Li, A.; Paudel, R.; Scholz, S.; Neumann, J.; Lees, A.; Revesz, T.; Hardy, J.; Mathias, C.J.; Wood, N.W.; et al. Glucocerebrosidase Mutations in 108 Neuropathologically Confirmed Cases of Multiple System Atrophy. Neurology 2009, 72, 1185–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caixeta, L.; Caixeta, V.D.M.; Nogueira, Y.L.; Aversi-Ferreira, T.A. Pharmacological Interventions in Corticobasal Degeneration: A Review. Dement. Neuropsychol. 2020, 14, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Kouri, N.; Whitwell, J.L.; Josephs, K.A.; Rademakers, R.; Dickson, D.W. Corticobasal Degeneration: A Pathologically Distinct 4R Tauopathy. Nat. Rev. Neurol. 2011, 7, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Saranza, G.M.; Whitwell, J.L.; Kovacs, G.G.; Lang, A.E. Corticobasal Degeneration. Int. Rev. Neurobiol. 2019, 149, 87–136. [Google Scholar] [PubMed]
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the Diagnosis of Corticobasal Degeneration. Neurology 2013, 80, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Kouri, N.; Ross, O.A.; Dombroski, B.; Younkin, C.S.; Serie, D.J.; Soto-Ortolaza, A.; Baker, M.; Finch, N.C.A.; Yoon, H.; Kim, J.; et al. Genome-Wide Association Study of Corticobasal Degeneration Identifies Risk Variants Shared with Progressive Supranuclear Palsy. Nat. Commun. 2015, 6, 7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Höglinger, G.U.; et al. Shared Genetic Risk between Corticobasal Degeneration, Progressive Supranuclear Palsy, and Frontotemporal Dementia. Acta Neuropathol. 2017, 133, 825–837. [Google Scholar] [CrossRef]
- Cali, C.P.; Patino, M.; Tai, Y.K.; Ho, W.Y.; McLean, C.A.; Morris, C.M.; Seeley, W.W.; Miller, B.L.; Gaig, C.; Vonsattel, J.P.G.; et al. C9orf72 Intermediate Repeats Are Associated with Corticobasal Degeneration, Increased C9orf72 Expression and Disruption of Autophagy. Acta Neuropathol. 2019, 138, 795–811. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Dystonia Locus | Chr Region | Gene Identified | Phenotype | Inheritance | Protein Name | Protein Function |
|---|---|---|---|---|---|---|
| Combined Dystonias (Dystonia-Plus Syndromes) | ||||||
| DYT3 | Xq13.1 | TAF1 | X-linked dystonia-parkinsonism | XR | Multiple transcript system, TAF1 | Core scaffold of transcription factor IID |
| DYT5a | 14q22.1–q22.2 | GCH1 | Dopa-responsive dystonia | AD | GTP cyclohydrolase 1 | Synthesis of tetrahydrobiopterin (BH4) |
| DYT5b | 11p15.5 | TH | Dopa-responsive dystonia | AR | Tyrosine hydroxylase (TH) | Synthesis of TH |
| DYT11 | 7q21–q31 | SGCE | Myoclonus-dystonia | AD | Epsilon-sarcoglycan | Unknown |
| DYT12 | 19q13 | ATP1A3 | Rapid-onset dystonia-parkinsonism | AD | Na(+)/K(+)-ATPase alpha3 subunit | Sodium pump |
| DYT15 | 18p11 | None | Myoclonus-dystonia | AD | N/A | N/A |
| DYT16 | 2q31.2 | PRKRA | Early-onset dystonia-parkinsonism | AR | Protein kinase | Stress response |
| - | 5p15.3 | SLC6A3 | Infantile parkinsonism-dystonia (dopamine transporter deficiency syndrome) | AR | Dopamine transporter (DAT) | Reuptake of dopamine from synapse |
| Isolated or Primary Torsion Dystonias (not Covered in this Review) | ||||||
| DYT1 | 9q34 | TOR1A | Generalized early-onset-limb dystonia | AD | TorsinA | Chaperone, ATP binding |
| DYT6 | 8p11.21 | THAP1 | Mixed-type dystonia | AD | THAP Domain Containing 1 | Regulates endothelial cell proliferation |
| DYT7 | 18p | None | Adult-onset cervical dystonia | AD | N/A | N/A |
| DYT13 | 1p36.13–36.32 | None | Craniocervical, laryngeal and limb dystonia | AD | N/A | N/A |
| DYT17 | 20p11.2–q13.12 | None | Familial dystonia | AR | N/A | N/A |
| DYT21 | 2q14.3–q21.3 | None | Adult-onset generalized or multifocal dystonia | AD | N/A | N/A |
| DYT25 | 18p11.21 | GNAL | Adult-onset cranial-cervical dystonia | AD | Guanine nucleotide-binding protein G(olf) subunit alpha | Signal transduction within the olfactory neuroepithelium and the basal ganglia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diez-Fairen, M.; Alvarez Jerez, P.; Berghausen, J.; Bandres-Ciga, S. The Genetic Landscape of Parkinsonism-Related Dystonias and Atypical Parkinsonism-Related Syndromes. Int. J. Mol. Sci. 2021, 22, 8100. https://doi.org/10.3390/ijms22158100
Diez-Fairen M, Alvarez Jerez P, Berghausen J, Bandres-Ciga S. The Genetic Landscape of Parkinsonism-Related Dystonias and Atypical Parkinsonism-Related Syndromes. International Journal of Molecular Sciences. 2021; 22(15):8100. https://doi.org/10.3390/ijms22158100
Chicago/Turabian StyleDiez-Fairen, Monica, Pilar Alvarez Jerez, Joos Berghausen, and Sara Bandres-Ciga. 2021. "The Genetic Landscape of Parkinsonism-Related Dystonias and Atypical Parkinsonism-Related Syndromes" International Journal of Molecular Sciences 22, no. 15: 8100. https://doi.org/10.3390/ijms22158100