1. Introduction
As ubiquitous messengers, calcium ions (Ca
2+) control the fate of the cells by regulating diverse Ca
2+-dependent pathways and transcription factors, including the nuclear factor of activated T-cells (NFAT) [
1]. Among the five members of these transcription factors, four (NFATc1-c4) are dynamically activated by the rise of cytoplasmic Ca
2+ [
2]. Under basal cytosolic Ca
2+ levels, NFAT proteins are heavily phosphorylated and trapped in the cytosol by masking the nuclear localization sequence (NLS) [
1]. However, upon intracellular Ca
2+ rise, the calmodulin-dependent phosphatase calcineurin is activated and dephosphorylates these transcription factors, exposing the NLS and inducing nuclear translocation [
1,
2]. Inside the nucleus, NFAT proteins initiate the transcription of several genes and, thereby, control the cell functioning in a Ca
2+-dependent manner. On the contrary, once the intracellular Ca
2+ concentration returns to the basal levels, re-phosphorylation of NFAT proteins by nuclear resident protein kinases leads to NFATs’ return into the cytosol by exposing the nuclear export sequence (NES) [
3].
Among other tissues, NFAT proteins are expressed in pancreatic β-cells [
4] and control the transcription of genes responsible for the proliferation and modulation of insulin secretion [
5,
6]. Notably, the source of Ca
2+ ions that drive NFAT proteins’ translocation to the nucleus differs among different cell types. In excitable cells, NFAT proteins are mainly activated via transmembrane Ca
2+ fluxes through the CaV1 family of L-type voltage-gated Ca
2+ channels [
7,
8]. However, activation of NFAT proteins via the L-type Ca
2+ channel requires a minimum threshold frequency of Ca
2+ oscillations [
9,
10]. Interestingly, not all members of the NFAT family are activated by the same frequency of Ca
2+ oscillations through the L-type Ca
2+ channel. For instance, only NFATc2 and NFATc3 proteins were shown to be activated upon glucose-induced Ca
2+ oscillations via L-type Ca
2+ channel in β-cells, indicating the isoform-specific sensitivity for Ca
2+ oscillations [
10].
It is reported that reactive oxygen species (ROS) production also induces NFAT translocation to the nucleus in various cell types [
11,
12]. Although the source of ROS-induced Ca
2+ oscillations differs among different cell lines [
13,
14,
15], the effect of ROS production on cytosolic Ca
2+ oscillations has been reported in multiple cell types [
13,
14]. In mast cells, it was reported that ROS production leads to cytosolic Ca
2+ oscillations via the phospholipase C (PLC)/inositol 1,4,5-trisphosphate (IP
3) pathway [
14]. In smooth muscle cells, Ca
2+ oscillations are generated through the L-type Ca
2+ channel [
13]. While exploring the effect of IP
3 generating agonist stimulation on NFATc3 translocation, we saw a strong induction of cytosolic Ca
2+ spiking and NFATc3 translocation by the near-UV light illumination usually applied for measuring cytosolic Ca
2+ with the most frequently used chemical Ca
2+ dye, Fura-2. Live-cell imaging revealed that near-UV light triggered ROS production, induces cytosolic Ca
2+ spiking via the L-type Ca
2+ channel and yielding NFATc3 translocation to the nucleus. Moreover, we present herein that the mitochondria’s ability to buffer cytosolic Ca
2+ makes them key regulators in controlling UV light-induced Ca
2+ spikes and NFATc3 translocation.
3. Discussion
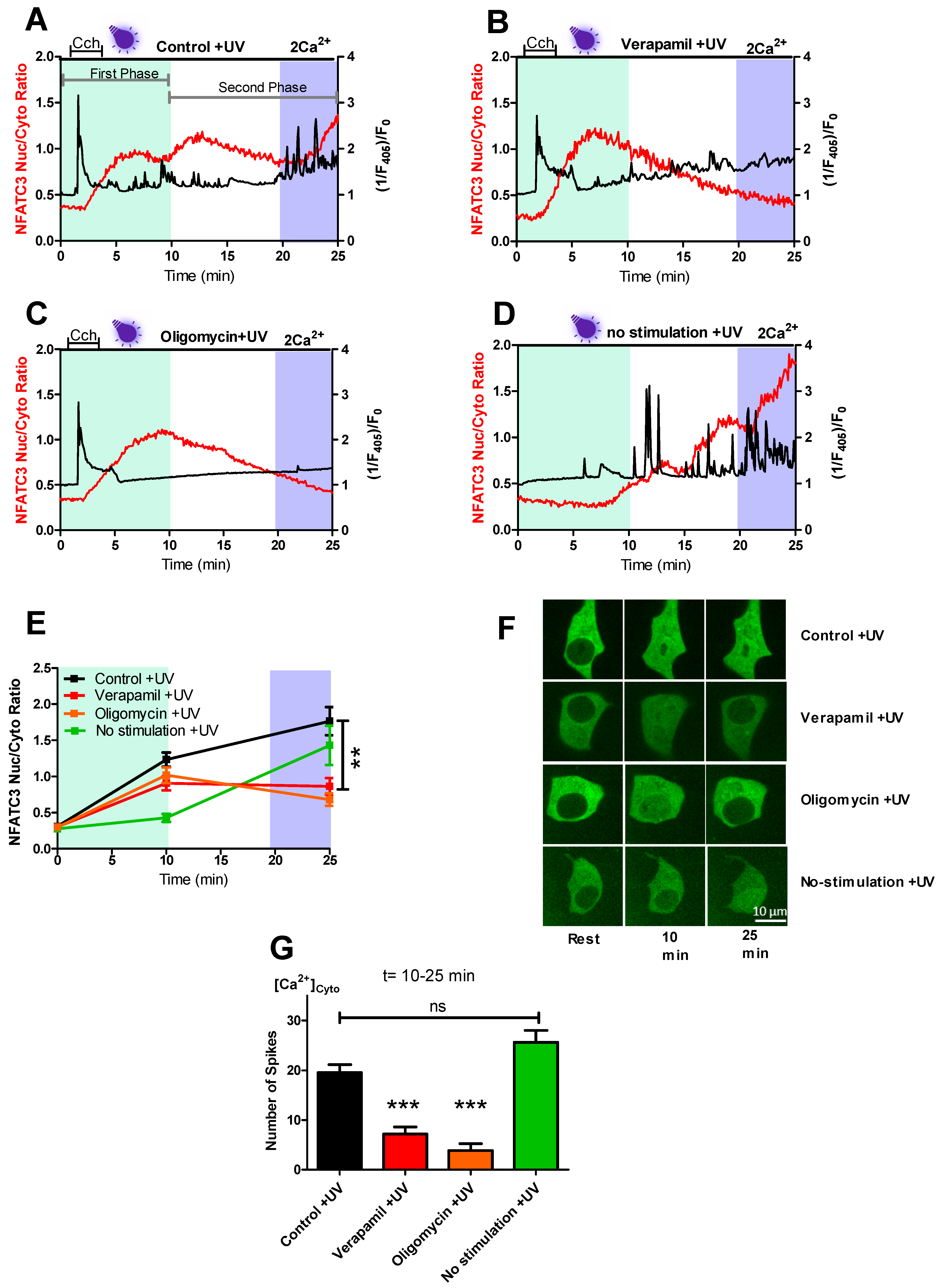
Among other NFAT members, NFATc3 has the highest expression level in human and mouse pancreatic islets [
5] and plays an important role in pancreatic β-cells [
10]. To gain more insight into the nuclear translocation mechanism of NFATc3, we investigated the translocation kinetics of NFATc3 in two distinct phases. In the first phase, CCh stimulation leads to intracellular Ca
2+ store mobilization, resulting in cytoplasmic Ca
2+ rise (
Figure 1A), which, in turn, activates NFATc3 migration to the nucleus. However, phase two was characterized by strong cytosolic Ca
2+ spikes leading to further migration of NFATc3 to the nucleus (
Figure 1A,E,F). Interestingly, the Ca
2+ spiking and NFATc3 translocation in the second phase were eliminated by verapamil treatment (
Figure 1B,G). In line with the previous study [
22], these results indicate that NFATc3 translocation in the first phase was the result of endoplasmic reticulum (ER) Ca
2+ release, which likely triggers Ca
2+ influx via calcium release-activated channels (CRAC) [
4]. In compliance with this assumption, verapamil treatment did not affect cytosolic Ca
2+ rise and NFATc3 translocation in the first phase but in the second phase.
Our findings that both the Ca
2+ spiking and NFATc3 translocation in the second phase were sensitive to mitochondrial ATP synthase inhibitor oligomycin are in agreement with the general expectation that the CCh-induced Ca
2+ release stimulates mitochondrial ATP production that, in turn, blocks the K
ATP channel and depolarizes the plasma membrane, thus promoting the opening of the L-type Ca
2+ channel and the subsequent nuclear translocation of GFP-NFATc3 (
Figure 1C,E–G). However, to our surprise, cells without CCh stimulation showed comparable repetitive Ca
2+ spiking and robust translocation of NFATc3 to the nucleus (
Figure 1D–G), thus eliminating the involvement of CCh stimulation on the Ca
2+ spiking and NFATc3 translocation during the second phase.
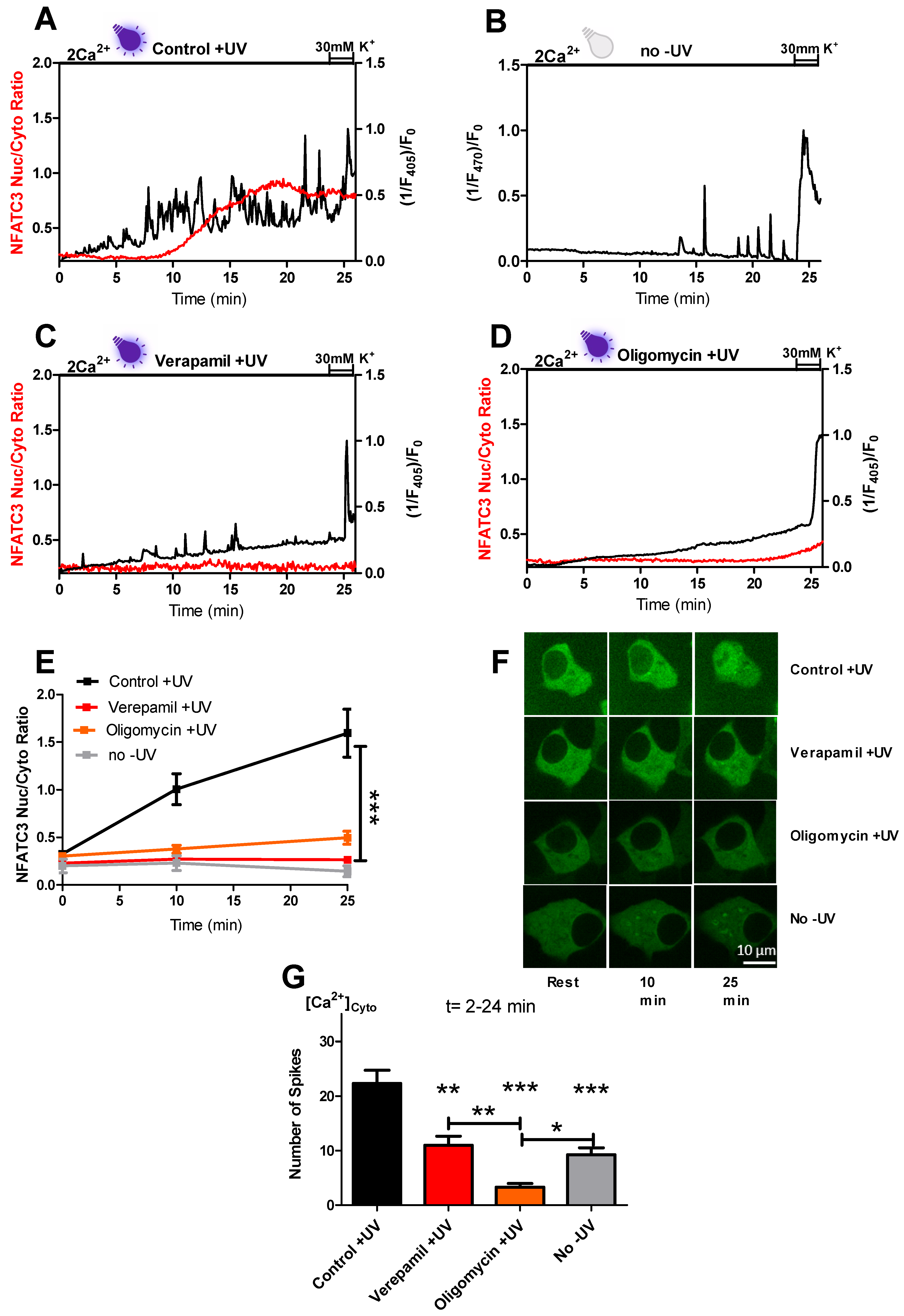
Therefore, we focused on an alternative trigger for cytosolic Ca
2+ spiking which drives NFATc3 translocation to the nucleus. In our spinning disc confocal microscopic setup, parallel measurements of cytosolic Ca
2+ using Fura-2 and GFP tagged NFATc3 translocation require the use of near-UV excitation with the high-energy 405 nm wavelength. We assumed that Ca
2+ spiking could be the result of near-UV light. Indeed, in the presence of near-UV light, we observed repetitive cytosolic Ca
2+ spikes which led to robust migration of NFATc3 to the nucleus (
Figure 2A,E–G). Previous studies support our results of near-UV light-induced cytosolic Ca
2+ oscillations and NFAT migration to the nucleus in different cell lines [
12,
14]. Since INS-1 cells show Ca
2+ spiking under basal conditions (
Figure 2B,G and
Figure S3a,f), we compared the amount of Ca
2+ spiking of cells exposed to near-UV light to non-UV stimulated control cells. Interestingly, Ca
2+ spiking in non-UV cells was halved compared to near-UV light-exposed cells (
Figure 2G). NFATc3 translocation to the nucleus did not occur in cells without near-UV light exposure (
Figure 2B,E–G), indicating that near-UV light is required to boost the Ca
2+ spikes for migration of NFAtc3 to the nucleus.
Furthermore, inhibition of L-type Ca
2+ channels with verapamil decreased the amount of UV light-induced Ca
2+ spiking to the level of non-UV used cells, and these Ca
2+ spiking were not enough to drive NFATc3 translocation to the nucleus (
Figure 2C,E–G). Thus, our findings confirm the published data on the impact of the frequency of Ca
2+ spiking on NFAT translocation to the nucleus [
9]. Additionally, these results highlight the source of near-UV light induced Ca
2+ spikes as well as the importance of a tight threshold in the amount of Ca
2+ spiking to fire NFATc3 migration to the nucleus. Next, we investigated whether basal ATP production is required to initiate near-UV light-induced cytosolic Ca
2+ spikes for NFATc3 translocation. Interestingly, oligomycin treatment eliminates the spiking as well as nuclear translocation of NFATc3, indicating that mitochondrial basal ATP production is indispensable for UV light-induced Ca
2+ spiking (
Figure 2D–G). However, the exact mechanism of ATP dependency of near-UV light-induced Ca
2+ spiking and NFATc3 translocation is not completely clear and needs further attention.
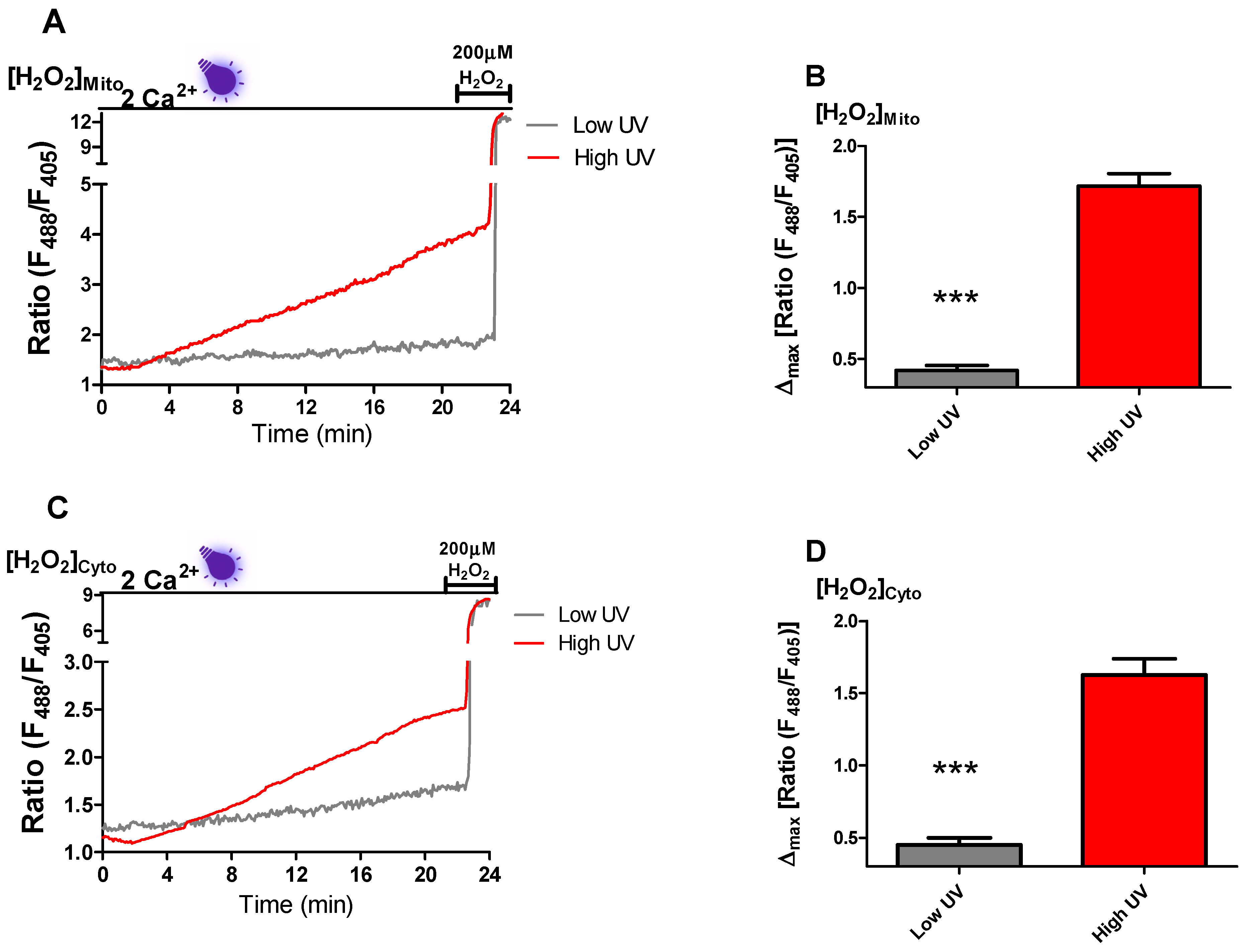
To shed light on the mechanism of near-UV light-generated Ca
2+ spiking, we checked mitochondrial and cytosolic ROS production in INS-1 cells. In line with a previous study [
14], both in mitochondria and cytosol, we observed a steady increase in ROS production in the presence of near-UV light (
Figure 3A–D). Surprisingly, near-UV light induced repetitive Ca
2+ spiking started mostly after 7 min of exposure, indicating that a certain threshold of ROS levels has to be reached to trigger L-type Ca
2+ channel activity and subsequent NFATc3 translocation. Interestingly, a former study indicated that moderate ROS levels induce β-cell proliferation [
23]. NFATc3 translocation to the nucleus increases the expression of genes responsible for cell proliferation [
5,
10]. It is tempting to speculate that β-cells have the necessity to accumulate a ROS threshold (e.g., during high metabolic activity) to activate NFATc3 driven proliferation.
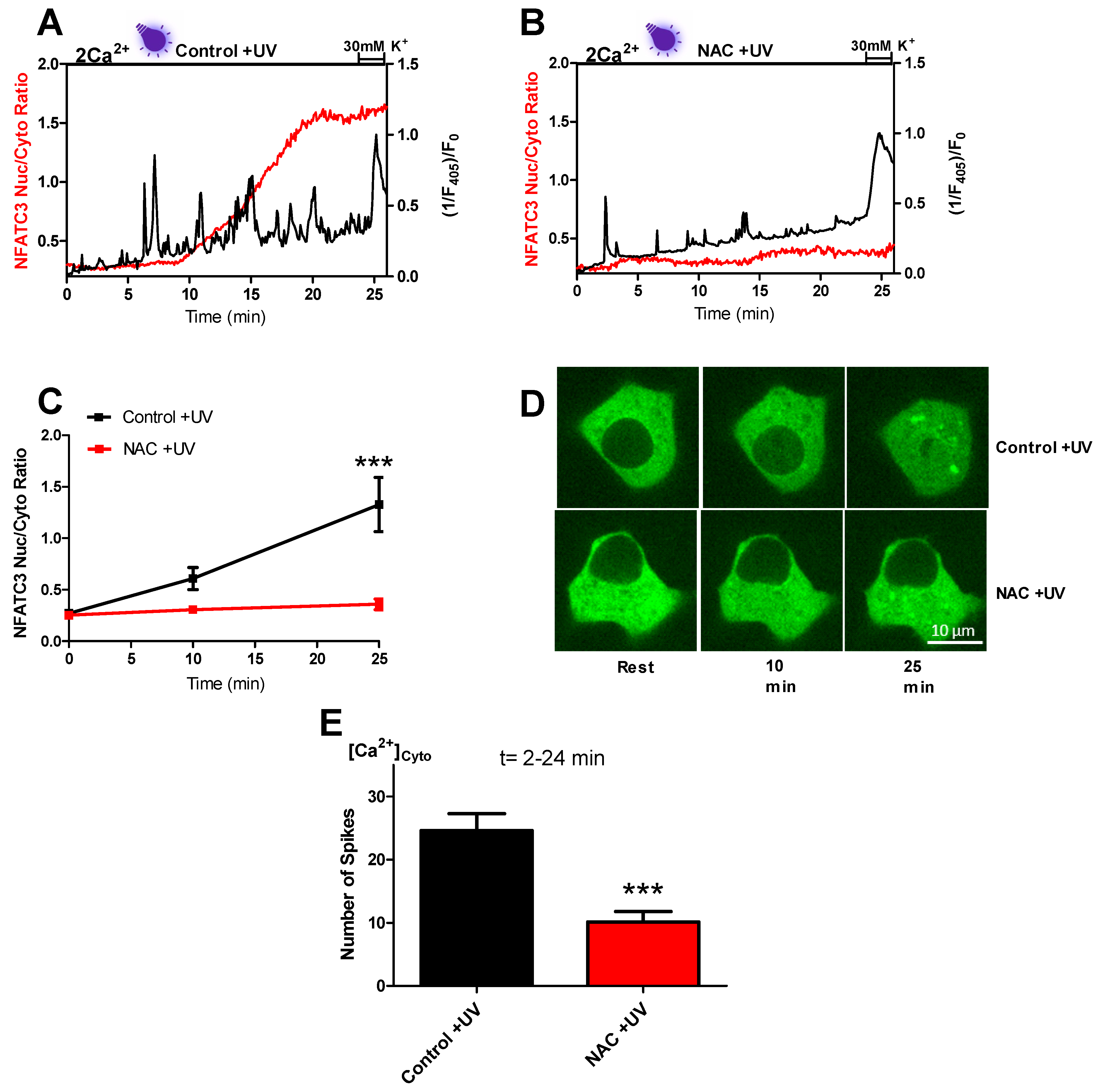
Migration of NFATc3 to the nucleus happens in a sequence of events that starts with near-UV light-induced ROS production. Consequently, we assumed that ROS scavenging prevents the signaling cascade. Indeed, NAC, a well-known ROS scavenger, decreased the amount of near-UV light-induced cytosolic Ca
2+ spiking (
Figure 4A,E). In support of our findings, another study also reported reduced UV light-generated cytosolic Ca
2+ oscillations upon NAC treatment [
14]. In response to hampered cytosolic Ca
2+ spikes, NFATc3 migration to the nucleus was also diminished by NAC treatment (
Figure 4B–E). These results support our assumption that near-UV light-induced ROS production leads to cytosolic Ca
2+ spiking through the L-type Ca
2+ channel, and these spikes drive NFATc3 translocation to the nucleus.
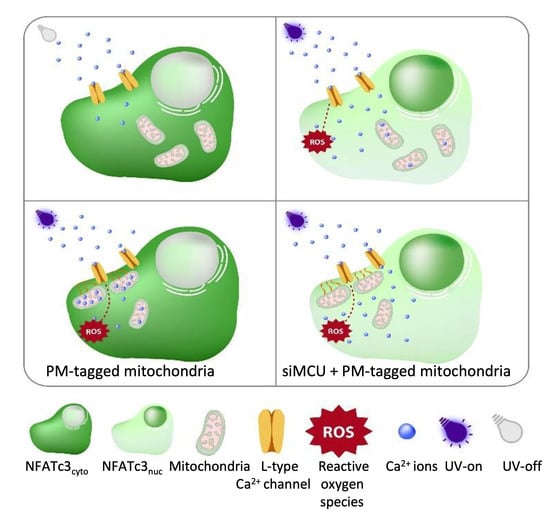
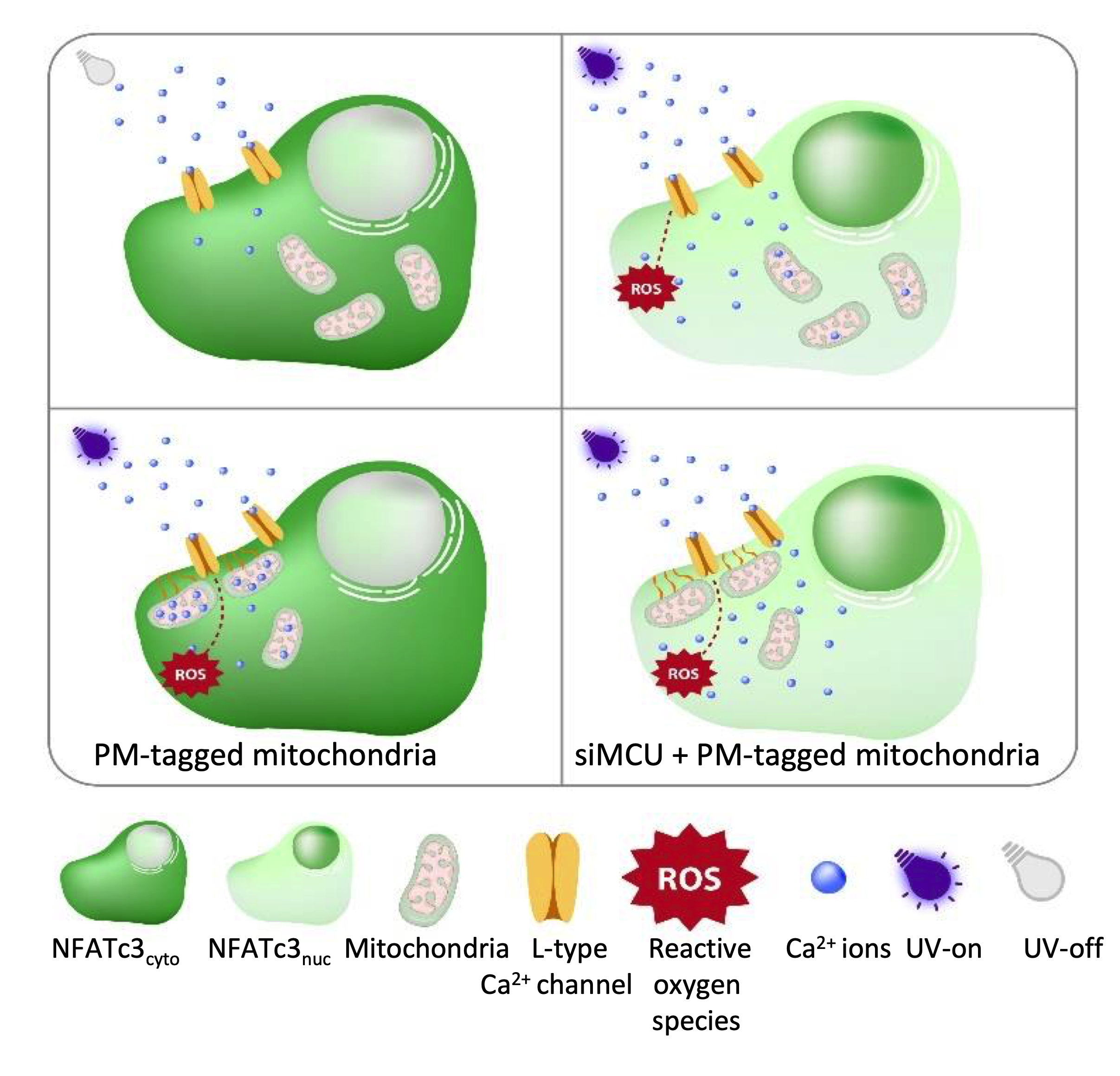
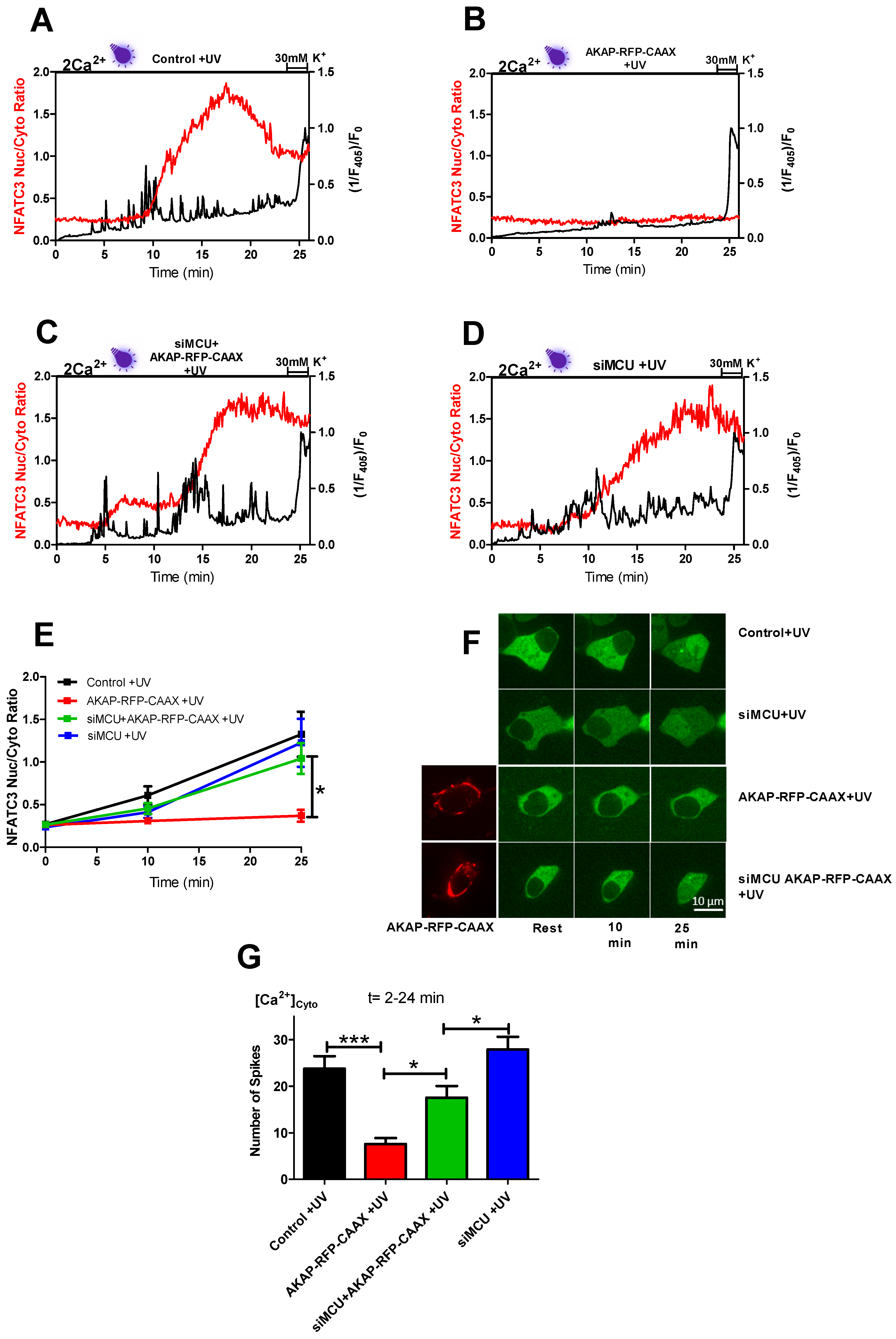
Having established that ROS induced repetitive cytosolic Ca
2+ spikes, which happen via the L-type Ca
2+ channels and drive the NFATc3 translocation to the nucleus, we wanted to distinguish between the direct effect of ROS and ROS-induced cytosolic Ca
2+ spiking on NFATc3 translocation. To do this, we came up with a hypothesis that if we buffer Ca
2+ spikes, which originate from L-type Ca
2+ channels at the plasma membrane, but keep the same ROS levels, we would not see the NFATc3 translocation and Ca
2+ oscillations. To test this idea, we positioned mitochondria to the sub-plasma membrane region using the AKAP-RFP-CAAX construct [
19,
20,
21] to buffer local Ca
2+ spiking. Interestingly, AKAP-RFP-CAAX-expressing cells did not alter ROS production (
Figure S4a–d) in response to near-UV light stimulation, but they eliminated near-UV light-induced Ca
2+ spiking as well as NFATc3 translocation (
Figure 5B,E–G), thus supporting our hypothesis that ROS-induced cytosolic Ca
2+ spiking, and not ROS itself, is required for NFATc3 translocation to the nucleus. To further validate this point, we knocked down MCU protein in AKAP-RFP-CAAX-expressing cells to diminish mitochondrial Ca
2+ uptake and buffering, which resulted in the reappearance of cytosolic Ca
2+ spiking and rescued migration of NFATc3 to the nucleus (
Figure 5C,E–G). A recently published paper revealed that 18% of the sub-PM area is occupied by mitochondria in β-cells [
24]. However, MCU KD alone did not affect UV light-induced Ca
2+ spiking and NFATc3 translocation (
Figure 5D–G). This could be because of the free motility of the sub-PM located mitochondria. The effect of near-UV induction on mitochondrial motility is not known. However, long-term intense blue light does not affect the sub-PM distributed mitochondrial density. In contrast, Ca
2+ entry via the L-type Ca
2+ channel significantly decreases the sub-PM located mitochondria from 18% to 13% in MIN6 cells [
24]. Thus, it is tempting to speculate that in control or siMCU cells, sub-PM localized mitochondria are not able to buffer UV light-induced Ca
2+ spiking to a great extent because of the Ca
2+ regulated mitochondrial redistribution.
In conclusion, this study reveals that near-UV light induced ROS production leads to repetitive cytosolic Ca2+ spiking via the activation of the L-type Ca2+ channel, triggering NFATc3 translocation to the nucleus. Moreover, we presented mitochondria as Ca2+ sinks by tagging them to the cell membrane, where mitochondria buffered cytosolic Ca2+ rises due to L-type Ca2+ channel activity and thereby prevented NFATc3 translocation. In addition, this study highlights the sensitivity of the pancreatic β-cells to near-UV light ranges frequently used in live-cell imaging for excitation of blue fluorescent proteins as well as cell-permeable dyes such as Fura-2.
4. Materials and Methods
4.1. Cell Culture and Transfection
The INS-1 832/13 (INS-1) cells were a generous gift from Prof. Dr. Claes B. Wollheim and Dr. Françoise Assimacopoulos-Jeannet (University Medical Center, Geneva, Switzerland). The INS-1 cells were cultured in RPMI 1640 containing 11 mM glucose (PubChem CID: 5793) supplemented with 10 mM HEPES (PubChem CID: 23831), 10% fetal calf serum (FCS), 1 mM sodium pyruvate (PubChem CID: 23662274), 50 μM β-mercaptoethanol (PubChem CID: 1567), 1% (v/v) Pen Strep® (ThermoFischer, Vienna, Austria; 10.000 U/L) 1.25 μg/mL Amphotericin B (ThermoFischer, Vienna, Austria; 250 μg/mL). Cells were used between passage numbers 53 and 68.
For all microscopic experiments, cells were plated on 30 mm glass coverslips in 6-well plates and transfected at 50–60% confluency with AKAP-RFP-CAAX (1 μg/well), GFP-NFATc3 (2 μg/well), mitoHyper7, and cytoHyper7 (1 μg/well) DNA constructs alone or with MCU siRNA (siRNA sequence: 5′-AAA GUC UCG UUU CGA CCU ATT-3′) by using 3 μL TransFast transfection reagent (Promega, Madison, WI, USA) in 1 mL of serum and antibiotic-free medium for 12–14 h. After that, transfection media was replaced with 2 mL of full culture medium. All experiments were performed 40-45 h after transfection.
4.2. Quantitative PCR
Total mRNA was isolated using the RNeasy® Mini Kit (Qiagen, Hilden, Germany), and reverse transcription was performed using Applied Biosystems High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific Baltics UAB, Vilnus, Lithuania). qPCR was performed using Promega GOTaq
® qPCR Master Mix (Madison, WI, USA). Knock-down efficiency of rMCU (
Figure S5) was determined using specific primers for rMCU (Forward: GCGTTGCCATCTATTCCCCA; reverse: TGGCTCAGGAGGTCTCTCTTT) and normalized to rGAPDH (Forward: TCTACATGTTCCAGTATGACTC; reverse: GCATCACCCCATTT GATG).
4.3. Buffers
Prior to experiments, cells were adjusted to room temperature with experimental buffer (EB): 2 mM Ca2+, 138 mM NaCl, 1 mM MgCl2, 5 mM KCl, 10 mM HEPES, 2.6 mM NaHCO3, 0.44 mM KH2PO4, amino acid and vitamin mix, 10 mM glucose, 2 mM L-glutamine, 1% penicillin/streptomycin, 1.25 μg/mL amphotericin B, pH adjusted to 7.4. All experiments were performed in following buffers 2CaNa buffer (2 mM CaCl2, 138 mM NaCl, 1 mM MgCl2, 5 mM KCl, 10 mM Hepes, 10 mM D-glucose, pH 7.4) and 2Ca30K buffer (2 mM CaCl2, 113 mM NaCl, 1 mM MgCl2, 30 mM KCl, 10 mM Hepes, 10 mM D-glucose, pH 7.4).
4.4. Live Cell Imaging Experiments
If not stated otherwise, all experiments were performed with a Zeiss array confocal laser scanning microscope (Axio Observer.Z1 from Zeiss, Gottingen, Germany) by using 100x objective lens (Plan-Fluor x100/1.45 Oil, Zeiss, Germany). This was equipped with a motorized filter wheel (CSUX1FW, Yokogawa Electric Corporation, Tokyo, Japan) on the emission side, an AOTF-based laser merge module for the 405, 445, 473, 488, 514, and 561 nm laser lines (Visitron Systems), and a Nipkow-based confocal scanning unit (CSU-X1, Yokogawa Electric Corporation). Data acquisition and control of the fluorescence microscope were performed using Visiview 4.2.01 (Visitron, Puchheim, Germany).
4.5. GFP-NFATc3 Translocation and Ca2+ Experiments with UV Light
GFP-NFATc3 translocation experiments were performed with the GFP-NFATc3 construct (gift from Mark L. Dell’Acqua, Department of Pharmacology, University of Colorado School of Medicine, Aurora). If not stated otherwise, all GFP-NFATc3 translocation experiments were performed using the following imaging parameters. GFP-NFATc3 was excited with 488 (30 mW) nm laser lines every 5 seconds for a 500-millisecond exposure time, and emissions were acquired at 510 nm. To stimulate the cells with UV light and follow the simultaneous changes in [Ca2+]cyto, cells were loaded with 3.3 µM Fura-2 in EB for 30 min and excited with 405 nm laser lines (20 mW) every 5 s for a 500-millisecond exposure time and emissions were acquired at 510 nm by using a charged CCD camera (CoolSNAP-HQ, Photometrics, Tucson, AZ, USA). VisiView acquisition software (Universal Imaging, Visitron Systems) was used to acquire the [Ca2+]cyto and translocation of GFP-NFATc3 from the cytosol to the nucleus. Background subtracted GFP-NFATc3 fluorescence ratio of the nucleus to cytosol region was used as a readout, where min 0 shows the resting ratio of GFP-NFATc3, min 10 shows the maximum translocation ratio of GFP-NFATc3 in the 0 to 10 min time interval whereas min 25 indicates the maximum translocation ratio of GFP-NFATc3 in the 20 to 25 min time interval.
To check bi-phasic GFP-NFATc3 translocation, Fura-2-AM loaded cells were stimulated with 100µM CCh in the presence of 2 mM extracellular Ca2+ and changes in [Ca2+]cyto and translocation of GFP-NFATc3 were recorded. The amount of cytosolic Ca2+ spiking was calculated between min 10 and 25.
Inhibition of [Ca2+]cyto spiking and GFP-NFATc3 translocation was performed by perfusing the cells in the presence of 2 µM oligomycin (Sigma-Aldrich, Vienna, Austria) and 10 µM verapamil (Sigma-Aldrich, Vienna, Austria) during the experiment or 30 min preincubation with 1 mM NAC (Sigma-Aldrich, Vienna, Austria). The number of cytosolic Ca2+ spikes was calculated between min 2 and 24.
To assess GFP-NFATc3 translocation without UV, cells were loaded with 3.3 µM Fura-2-AM in EB for 30 min. In the presence of 2 mM extracellular Ca2+, a 488 nm excitation laser was used to track GFP-NFATc3 migration alone for 26 min.
4.6. Analysis of Cytosolic Ca2+ Spiking
First, cytosolic Ca2+ traces were background corrected using background ROI intensities. Furthermore, the standard deviation (SD) of basal Ca2+ signals was calculated to estimate the signal-to-noise ratio. First, peaks were identified by a change from positive to the negative slope between measurement points. As a second characteristic, intensity changes between time points of positive to negative slope changes and negative to positive slope changes had to exhibit 5-fold higher values than the basal SD values to be counted as a valid signal and identified as a Ca2+ spike.
4.7. Mitochondrial and Cytosolic ROS Measurements
Mitochondrial and cytosolic ROS measurements were performed with the recently developed genetically encoded H
2O
2 sensors mitoHyper7 and cytoHypher targeted to the matrix or cytosol, respectively [
17]. Both sensors were excited with 405 and 488 nm laser lines, and emissions were acquired at 510 nm. To record the baseline, cells were excited at 5 mW with 405 and 488 nm laser lights every 5 seconds with a 500-millisecond exposure for 2 min. At the end of min 2, excitation with the 405 and 488 nm lasers was increased to 20 milliwatts and measurement was performed for 22 more min in these settings. At the end of min 22, cells were stimulated with 200 µM of H
2O
2 to obtain the saturation ratio of the sensors. Control cells were excited with 5 mW 405 and 488 nm laser lights every 5 seconds with 500-millisecond exposure for 22 min where ROS production did not occur. The background subtracted maximum UV light-induced H
2O
2 ratio of 488/405 was subtracted from the baseline to obtain the change in ROS production at the end of 20 min with 20 mW repetitive UV induction.
4.8. Cytosolic Ca2+ Measurements without UV Light
Cytosolic Ca
2+ measurement without UV light was performed with an Olympus IX73 inverted microscope. This microscope was equipped with an UApoN340 40× oil immersion objective (Olympus, Tokyo, Japan) and a CCD Retiga R1 camera (Q-imaging, Vancouver, BC, Canada). LedHUB
® (Omnicron, Rodgau, Germany) equipped with 340, 385, 455, 470, and 550 nm LEDs. A GFP (GFP-3035D, Semrock, Henrietta, NY, USA) filter set was used for illumination of Fluo-4. Visiview 4.2.01 (Visitron, Puchheim, Germany) was used for the data acquisition. Alternatively, an AnglerFish F-G/O (Next Generation Fluorescence Imaging/NGFI (
www.ngfi.eu), Graz, Austria) was used for data acquisition. Subsequent data analysis was performed in ImageJ (NIH, Bethesda, MD, USA) and Excel (Microsoft, Redmond, WA, USA). Both microscopes were equipped with an automatic perfusion system PS-9D (NGFI).
To avoid usage of UV light, cells were loaded with 3.3 µM Fluo-4 in EB for 30 min. On the microscope, cells were perfused with 2 mM Ca2+ buffer for 24 min and 2-min 30 mM K+ buffer to get maximum uptake through the L-type Ca2+ channels. The depolarization induced maximum Ca2+ uptake via L-type Ca2+ channel is used for the normalization of Fura-2. The number of cytosolic Ca2+ spikes was calculated from min 2 to min 24.
4.9. Data Analysis
The data shown above were acquired from three different days and represent the mean ± SEM. The number of single cells is represented as n = cell number in each figure legend. Single cells were used for the statistical analysis, where Student’s t-test and analysis of variance (ANOVA) with Tukey post hoc test were performed. GraphPad Prism software version 5.04 (GraphPad Software, San Diego, CA, USA) and Microsoft Excel (Microsoft) were used for the analysis, calculation, and representation of the data.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




