1. Introduction
Proteins are present in many biological processes in the cells of living beings, playing different functions, such as transport, growth, and maintenance of the body. They are formed by a sequence of amino acids, which consist of the protein’s primary structure [
1]. Amino acids interact physically and chemically with each other, forming three-dimensional structures. The local three-dimensional structure that each amino acid participates in is called the secondary structure, whereas three-dimensional structure that the protein forms is called the tertiary structure [
2].
Analyzing the three-dimensional structures of proteins has a great impact on determining the protein functions [
3], mainly because each function depends on a specific folding [
4], and each protein can exert more than one function [
5], as well as the development of new applications, such as drug and enzyme design and biosensors [
6,
7,
8]. Recent work [
9] has shown that the prediction of the tertiary structure, directly from the primary structure, can achieve good results, but this task is still open. The most common method in the literature is to first understand the secondary structure and then predict the tertiary structure.
To determine the protein structures, laboratory methods are applied, such as X-ray crystallography, multidimensional nuclear magnetic resonance, and infrared spectroscopy [
10], with a complex interpretation of the results. For instance, to determine the secondary structures of the protein, it is necessary to analyze the hydrogen bonding patterns and geometric constraints, and to use the DSSP tool [
11]. On the other hand, the number of sequenced proteins grows faster each year, due to advances in gene sequencing [
12].
Figure 1 shows the number of proteins sequenced on UniProtKB [
13], the main repository of sequenced proteins, and PDB [
14], the main repository of proteins with three-dimensional structures defined by laboratory methods, such as secondary and tertiary structures. Thus, computational methods have become important to help predict protein secondary structures efficiently and effectively.
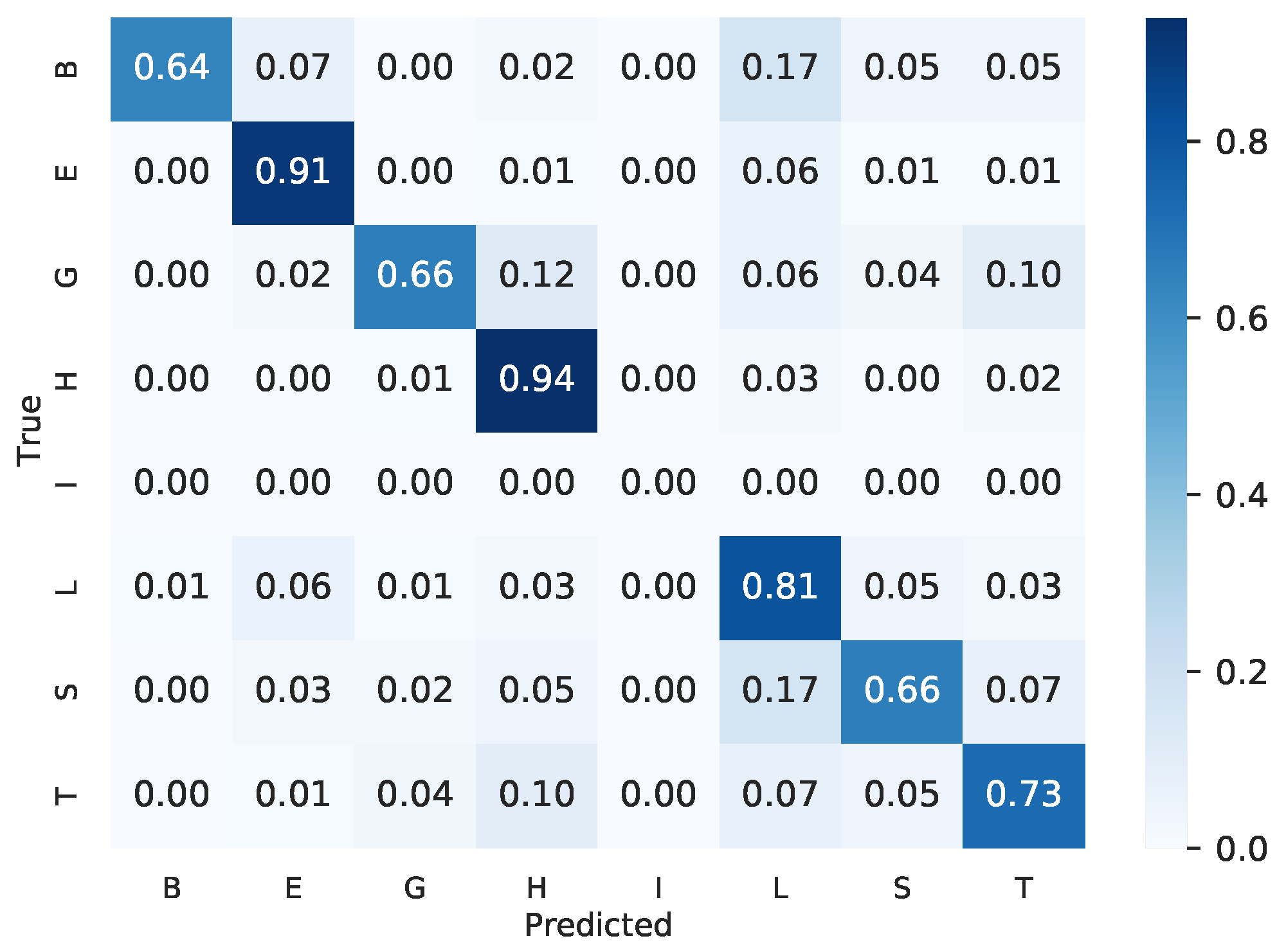
The protein secondary structure prediction task considers that each amino acid can fold into one of eight possible classes. These classes are “B” (residue in isolated beta bridge), “E” (extended strand), “G” (3-helix), “H” (alpha helix), “I” (5-helix), “L” (loop), “S” (bend), and “T” (hydrogen-bonded turn) [
3].
The protein secondary structure prediction classifiers in the literature are divided into template-based and template-free classifiers. The template-based classifiers use tools to search and find local alignments between proteins [
15], which are usually employed by the Basic Local Alignment Search Tool (BLAST) [
16]. The template-free classifiers apply machine learning and deep learning techniques to learn patterns and predict the structures from unseen proteins.
The prediction methods using template-free classifiers for this task started in the 1970s, after Chou and Fasman’s work [
17], mainly using statistics and manual rules [
18,
19]. These first methods for predicting secondary structures of proteins were evaluated in small databases, due to the lack of computational power and the amount of proteins with secondary structures defined by laboratory methods at that time. Furthermore, as they are methods that use simple analyses, most of them were created from a few samples and by manual processes; these methods are less effective than the methods of later decades.
With the development of computer power, the growth of databases, such as PDB [
14], CB513 [
20], and the biennial Critical Assessment of protein Structure Prediction (CASP) [
21], new template-free methods have become popular, such as simple multilayer perceptron neural networks [
22,
23,
24], support vector machines [
25], clustering [
26], and hierarchical classifiers [
27]. At this phase of the problem, of predicting protein secondary structures, the methods employed sliding window techniques to perform the classifications. However, the classifiers did not show a consensus regarding the optimal size of the window used. Furthermore, as these methods can only analyze the local window of interaction between amino acids, longer interactions—that is, amino acids at distant positions in the sequence—are not verified, and can be important to determine the secondary structures.
After 2010, template-free classifiers became the state-of-the-art, mainly using deep learning techniques, such as recurrent neural networks (RNNs) with gated recurrent unit (GRUs) [
28] and long short-term memory (LSTM) [
29] modules, convolutional neural networks (CNNs), and ensemble techniques. RNN classifiers [
30,
31,
32] receive the entire sequence as input, being able to analyze the chain globally, surpassing the methods that use sliding windows. However, the main negative for this approach involves the computational cost of LSTM and GRU neurons, mainly in bidirectional classifiers, in addition to the problem of vanishing and exploding the gradient.
CNN classifiers [
33,
34,
35] have filters that can analyze several different windows of a sequence. Deep convolutional networks can obtain long-distance information, but local information, gained in the early layers of the network, is lost. With that, several methods have started to use inception blocks, where information from the shallower layer is kept, concatenating the information with the deeper layers. This approach has enabled an advance in the results of the prediction of secondary structures, such as the method developed by Ratul et al. [
33], which reached the state-of-the-art in CB6133 [
7] and CB513 [
20] datasets, with a classifier with inception blocks.
The ensemble techniques for classifiers have shown that they can achieve better results than individual classifiers. Oliveira et al. [
12] presented a method for making the ensemble techniques for classifiers, using a bag of bio-inspired optimization algorithms to find weights for each class of each classifier. The authors showed that the genetic algorithm has a greater impact on the final result than the other optimization algorithms. Drori et al. [
36] combined several classifiers, using the highest mean prediction value of the classifiers. The approach presented by Drori et al. [
36] is penalized by outliers; that is, classifiers that are not ’very sure’ can harm the classification result. Kumar et al. [
1] presented a method that uses both RNN and CNN to perform the classification, using CNN as a local classifier and RNN as a global classifier.
Regarding template-based classifiers, BLAST [
16] was created in the 1990s, as well as methods using this tool to make predictions of protein secondary structures [
12,
37,
38]. However, template-based methods are not as explored in the literature as template-free classifiers.
Ensemble methods that use evolutionary optimization algorithms have been used in several domains. Haque et al. [
39] used a genetic algorithm to find the best combination of algorithms for Alzheimer’s disease classification and face recognition. The method finds the best combination of classifiers, and the fusion is conducted with a simple voting strategy. Prado et al. [
40] used a genetic algorithm to find the best combination of classifiers for the energy consumption forecasting problem, while Kausar et al. [
41] and Aburomman and Reaz [
42] applied the particle swarm optimization algorithm to perform weighted voting of multiple classifiers to predict data from the UCI repository and for detection of TCP/IP connections, respectively. Kardas et al. [
43] also employed optimization by evolutionary algorithms for the classification of data from the UCI repository, but unlike the methodology of Kausar et al. [
41], the proposed method used genetic algorithms.
The methods used by Prado et al. [
40], Kausar et al. [
41], Aburomman and Reaz [
42], and Kardas et al. [
43] found weights for each classifier. These approaches can be disadvantageous if a classifier correctly classifies a class and receives a lower weight in the voting system or a weight equal to 0. Dimilliler et al. [
44] compared this methodology with the use of weights equal to 1 or 0 for each class of each classifier during the prediction, making the ensemble using the weighted voting per class and obtaining the highest value. To find the weights, their method applied genetic algorithm. As a result, Dimilliler et al. [
44] demonstrated that their approach can achieve better results than finding unique weights for each classifier during the ensemble for a named-entity recognition task of biomedical paper abstracts. Parvin and Alizadeh [
45] showed that, by giving weights to each class of each classifier from 0 to 1, the ensemble can reach better results than binary weights, as in the method proposed by Dimilliler et al. [
44], with experimental evidence for classification of Farsi digits. Ekbal and Saha [
46] also used the genetic algorithm to find weights for each output of each classifier for the weighted voting for the named-entity recognition problem for Indian languages.
In the present study, we investigate six different template-free classifiers and two different template-based classifiers for protein secondary structure prediction. Our template-free classifiers have both local classifications, made with the sliding windows technique (random forest classifier), and global classifiers (RNN classifiers). We also evaluate inception blocks (inception-v4 blocks classifier), inception blocks with GRU recurrent layers (inception recurrent network classifier), as well as classifiers for specific characteristics of the database (a BERT-based classifier and CNN classifier). For the template-based classifiers, we applied a configuration that uses only the best local alignments, but cannot predict all structures for all amino acids, and a general configuration, which can predict for all amino acids.
In addition, we explore the ensemble among template-based classifiers, as well as the ensemble of template-free and template-based ensembles using genetic algorithm optimization. The proposed optimization algorithm finds weights for each class of each classifier. As our main contribution, we explore different representations of the protein features, making our classifiers complementary, obtaining results that surpass the state-of-the-art approaches by 8.2 percentage points on the CB6133 test set and 17.6 percentage points on the CB513 test set.
3. Discussion
The objective of this study was to investigate different classifiers with distinct views of the representation of the protein features. Our method has two main ways to make the prediction—template-free and template-based classifiers. Both of them surpassed the state-of-the-art approaches on the CB6133 and CB513 datasets.
For the template-free classifiers, the ensemble between them showed that the most important classifier was BERT, which was the predictor that received the highest weights in more classes. This may have been related to the recent success of this type of algorithm, based on transformers, in different tasks, as in natural language processing (NLP), as shown by BERT, and for image classification, as shown by vision transformer [
55].
The results achieved by our template-free classifiers are competitive with the results obtained by other classifiers in the literature. We conjecture that, with more data, our method can obtain better Q8 accuracy because deep learning algorithms have more generalization capability under a variety of data. Further investigation of different types of data augmentation, as augmentation for images, for text, and for tabular data, specific for each one of our classifiers, may help to achieve better results.
The main drawbacks of all of our template-free classifiers involve the lack of explainability of the results; that is, understanding why each classifier makes the prediction of one class and the computational cost of the whole method, which includes training all of the classifiers of one type of classifier (for instance, training ten different bidirectional recurrent networks that compose the RNN classifier), to make the ensemble between this classifier and the template-free classifier. One example of these drawbacks is related to random forest. This classifier is the easiest to verify the decisions made, however, there are 2500 trees to manually verify and understand the choices, making this infeasible.
Our template-based classifier had the highest Q8 accuracy between the basic ensembles (template-free and template-based). We believe that this classifier achieved better results due to the high similarity of the proteins in the test set with the proteins with secondary structures determined by the laboratory methods of PDB. Future works can investigate the impact of limited search databases on the prediction of secondary structures.
The results of the ensemble evaluation section demonstrate that our ensemble method (GA-based) can achieve better results compared to the meta-classifiers. The investigation of other types of ensemble techniques, using deep features, can be a path for future work.
4. Materials and Methods
In this section, we describe our template-free and template-based classifiers for predicting protein secondary structures, as well as the ensemble technique employed. We also present the evaluation metrics and the datasets employed in our work.
4.1. Template-Free Classifiers
In this subsection, we present our six template-free classifiers—bidirectional recurrent neural network, random forest, inception-v4 blocks, inception recurrent network, BERT, and convolutional neural network.
The execution time required to train each classifier to make predictions on the CB6133 and CB513 datasets, as well as to ensemble the template-free classifiers, is shown in
Table 22. In all experiments, we used the Google Colaboratory infrastructure (
https://colab.research.google.com, accessed on 22 October 2021).
4.1.1. Bidirectional Recurrent Neural Networks
Recurrent neural networks (RNN) are capable of analyzing and classifying sequences based on past information. However, for the protein secondary structure prediction task, the “past” (the predecessor amino acids) and the “future” (the successor amino acids) impact the secondary structure of an amino acid. Therefore, bidirectional recurrent neural networks are more effective for this task. Even more, this type of classifier can deal with the whole sequence, belonging to the set of global classifiers.
Based on this, we evaluated the use of bidirectional recurrent networks for this task. We analyzed different configurations of RNNs, varying the number of bidirectional layers and the number of neurons per layer. Due to the smaller number of parameters than the LSTM [
29], we performed the experiments using GRU [
28] memory modules. The best configurations were using 600 neurons per layer and {2, 3, 4, 5, 6} layers.
As the amino acid sequences are sparse vectors, this is 20 features equal to 0 and 1 feature equal to 1, such as one-hot encoding, we evaluated the presence of an embedding layer for this information. The remaining features, the position-specific scoring matrix (PSSM) information, go directly to the first bidirectional recurrent layer. The results on the validation set showed that this approach helped improve the results.
Figure 4 shows the general configuration of our RNN classifier.
For each configuration, we analyzed the ensemble of two networks with the same configuration, with one of them evaluating the sequence in the standard direction, this is in the form that the sequences are present in the files, and another analyzing the inverted way, i.e., from the end to the beginning of the sequence. The probabilities of the two networks are concatenated and normalized, with the sum of all eight classes for each amino acid equal to 1. The final result of the ensemble of each configuration is considered the final prediction of that configuration.
We trained all networks with the TensorFlow [
56] framework by 50 epochs, with a learning rate equal to
, categorical cross-entropy loss function, Adam optimizer, early stopping, and reducing the learning rate on plateau by
. In the end, we made the ensemble between the five configurations and considered this final prediction as the prediction of the RNN classifier.
4.1.2. Random Forest
The local interaction between close amino acids—different from the global analysis made by RNN—also has an impact on the prediction of protein secondary structures. Between the local interaction classifiers, the methods in the literature usually use sliding window-based predictors; however, each one of them employed different sliding window sizes.
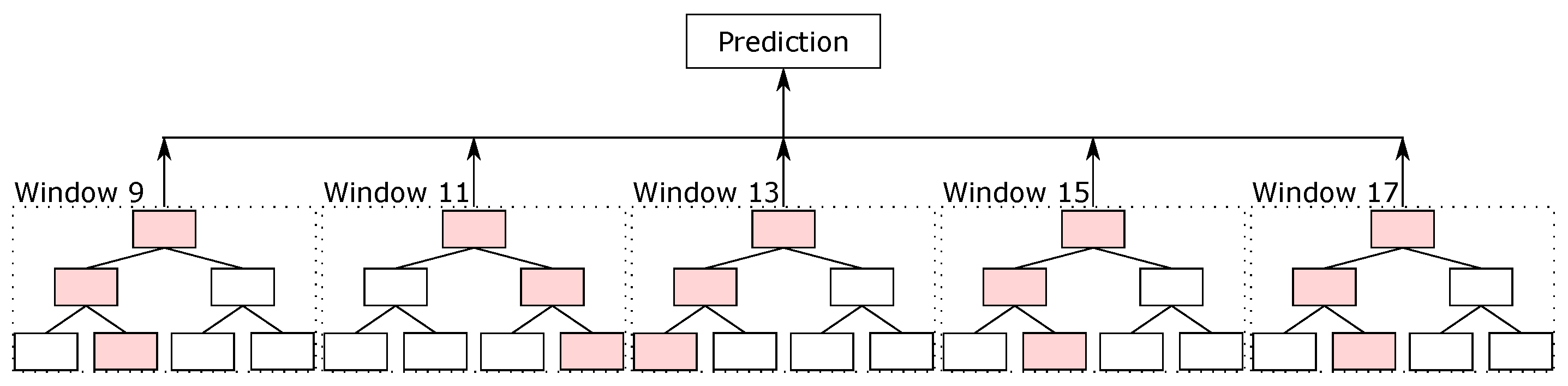
Driven by the local classifiers available in the literature for the protein secondary structure prediction task, we created our local classifier based on random forest. In this classifier, we split the sequence into blocks, and the classifier predicted the secondary structure of the central amino acid. We evaluated different parameters, such as the number of trees, the maximum depth of the trees, and the window sizes, on the validation sets. The best parameters that we found were 500 trees per random forest classifier, with the maximum depth equal to 15, and with 5 different window sizes, from 9, i.e., 4 amino acids before and 4 after the central amino acid, up to 17 (that is, windows of sizes 9, 11, 13, 15, and 17). For padding, we used the same idea as used in the databases—values equal to 0 at the beginning and end of the sequences.
With the five random forest classifiers with different sliding window sizes, we made the ensemble between them. The final result made by the ensemble is considered the final prediction of random forest, which we called the RF prediction.
Figure 5 illustrates the ensemble of random forest classifiers.
4.1.3. Inception-v4 Blocks
Convolutional neural networks can obtain local information from images from the first layers of the architecture. When the network becomes deeper, the local information gets lost, mainly, due to that, deep layers can understand global and general features of images.
The recent results of inception-based [
57,
58] protein secondary structure prediction methods [
33,
49,
51] showed that this kind of architecture can aggregate local information, generated by the first layers, and global information, which is generated by deep layers. Driven by this success, we evaluated the newest inception architecture (Inception-v4 [
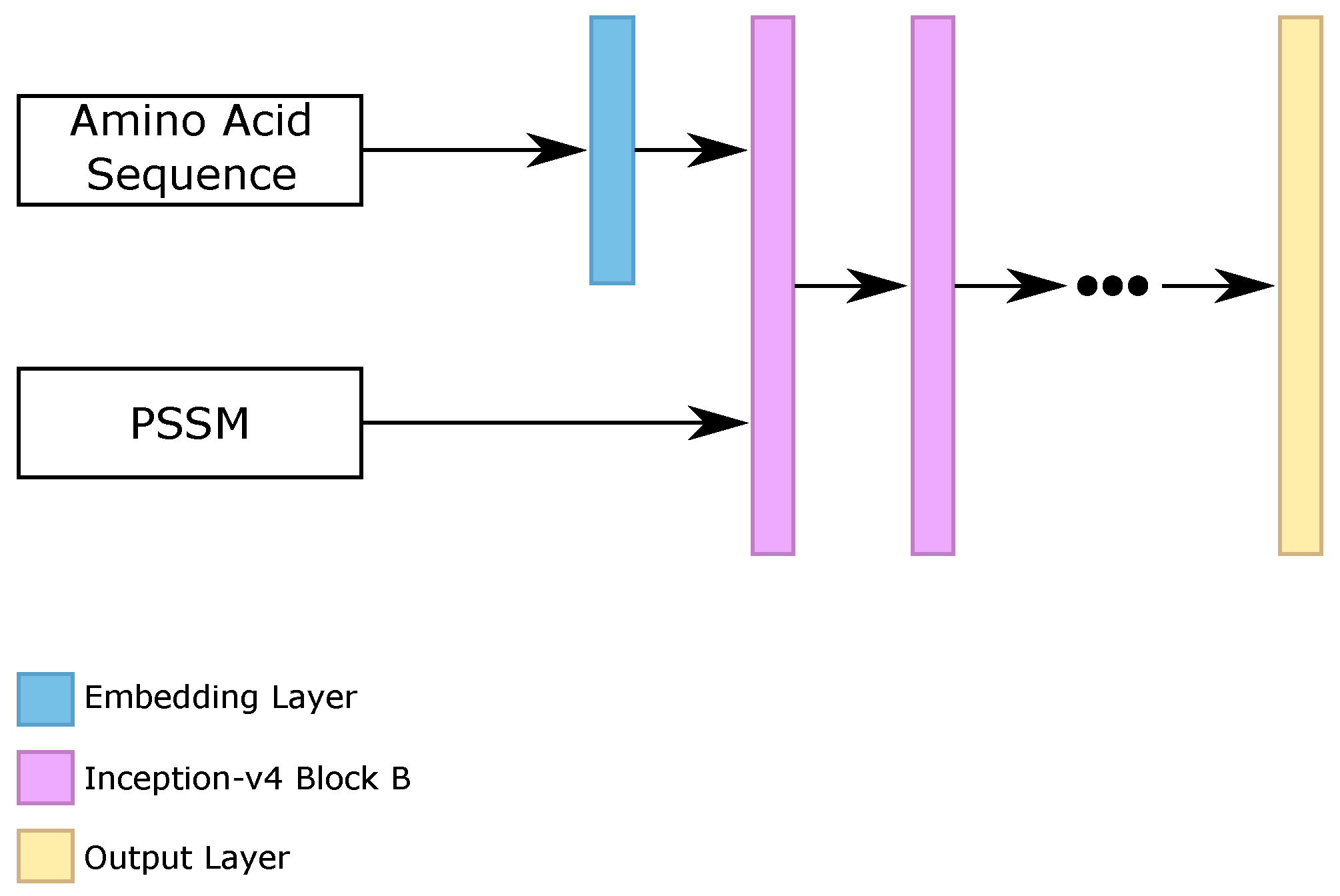
58]) for the protein secondary structure prediction task. As the original inception-v4 was created to make image classification, using 2D convolutions and 2D poolings, we transformed the 2D convolutions and 2D poolings into 1D operations, because our task consisted of analyzing sequences of amino acids, instead of analysis of image parts.
We evaluated the three blocks that composed the inception-v4 architecture, called “block A”, “block B”, and “block C”, stacking from 1 to 10 blocks of the same type. Moreover, we analyzed the use of embedding layers for the sparse amino acid sequence, as we did on the RNN classifier. The best five configurations found on the validation set were using {3, 4, 5, 6, 7} stacked “blocks B”, using the embedding layer for the amino acid sequence.
We trained all networks with TensorFlow [
56] framework for 50 epochs, with learning rate equal to
, categorical cross-entropy loss function, Adam [
59] optimizer, early stopping, and reducing learning rate by
after five epochs, without better results on the validation set.
Figure 6 illustrates the general architecture of our inception-v4 block (Iv4B) classifiers.
After the prediction of the five different classifiers (that we called inception-v4 block (Iv4B), we made the ensemble between them. We considered the final ensemble as the prediction of the Iv4B classifier.
4.1.4. Inception Recurrent Networks
Driven by the development of our Iv4B classifier, we evaluated the stacking of bidirectional recurrent layers after the stacked inception-v4 blocks. Our main goal with this method was to use the representations learned by Iv4B classifiers as input to a purely global classifier, in this case, bidirectional recurrent networks.
We employed the five variations of Iv4B, and we evaluated a different number of bidirectional recurrent layers (1, 2, 3, 4, 5), and the number of neurons per layers (100, 200, 300, 400, 500). Our best results on the validation set used 3 bidirectional recurrent layers, with 100 neurons per layer.
We trained all of the networks with TensorFlow [
56] framework for 50 epochs, with learning rate equal to
, categorical cross-entropy loss function, Adam [
59] optimizer, early stopping, and reducing learning rate by
after five epochs, without better results on the validation set.
Figure 7 illustrates the general architecture of this classifier.
Then, we made the prediction between the five configurations of this classifier (that we called inception recurrent networks, IRN). The ensemble is considered the prediction of the IRN classifier.
4.1.5. BERT
Transformer-based [
60] architectures achieved good results in many natural language processing (NLP) tasks. The most famous architecture based on transformers is the Bidirectional Encoder Representations from Transformers (BERT) [
61]. This architecture surpassed the state-of-the-art approaches in many NLP tasks, such as sentiment analysis, named-entity recognition, sentiment analysis, and text classification. Based on the recent success of BERT, many methods for biological processes began to use BERT for different applications [
62,
63,
64]. Furthermore, as BERT was originally trained on English texts, a new version of BERT became popular for proteins, which was trained on the BFD [
65] dataset. This new version, called BERT-prot [
64], showed good results on different tasks based on protein classifications.
Driven by this success of BERT-prot on protein classification problems, we evaluated two different views for the protein secondary structure prediction application. In the first one, we applied the BERT-prot for the named-entity recognition task. In this type of classification, each amino acid is classified in one of each entity, or secondary structure. This first classifier uses all of the sequences as input, so it can analyze the protein as a global classifier. For the second view, we transformed the protein secondary structure into local classification, i.e., we broke the sequences into parts and used each part as a text classification task, classifying the central amino acid. In these two different views, we only used the letters that represented each amino acid and excluded the PSSM information. For text classification, we created padding using special characters.
Figure 8 shows all six BERT classifiers.
For the text classification task, we analyzed different window sizes, and the best results on the validation set were obtained by five different configurations (21, 41, 61, 81, 101). All classifiers were fine-tuned by five epochs, using the ktrain [
66] package, Adam [
59] optimizer, learning rate equal to
, and early stopping.
In the end, we had six classifiers (one using the named-entity recognition task view, and five using the text classification task view). Then, we made the ensemble between them. The ensemble of BERT-based classifiers was labeled as ’the BERT classifier’.
4.1.6. Convolutional Neural Networks
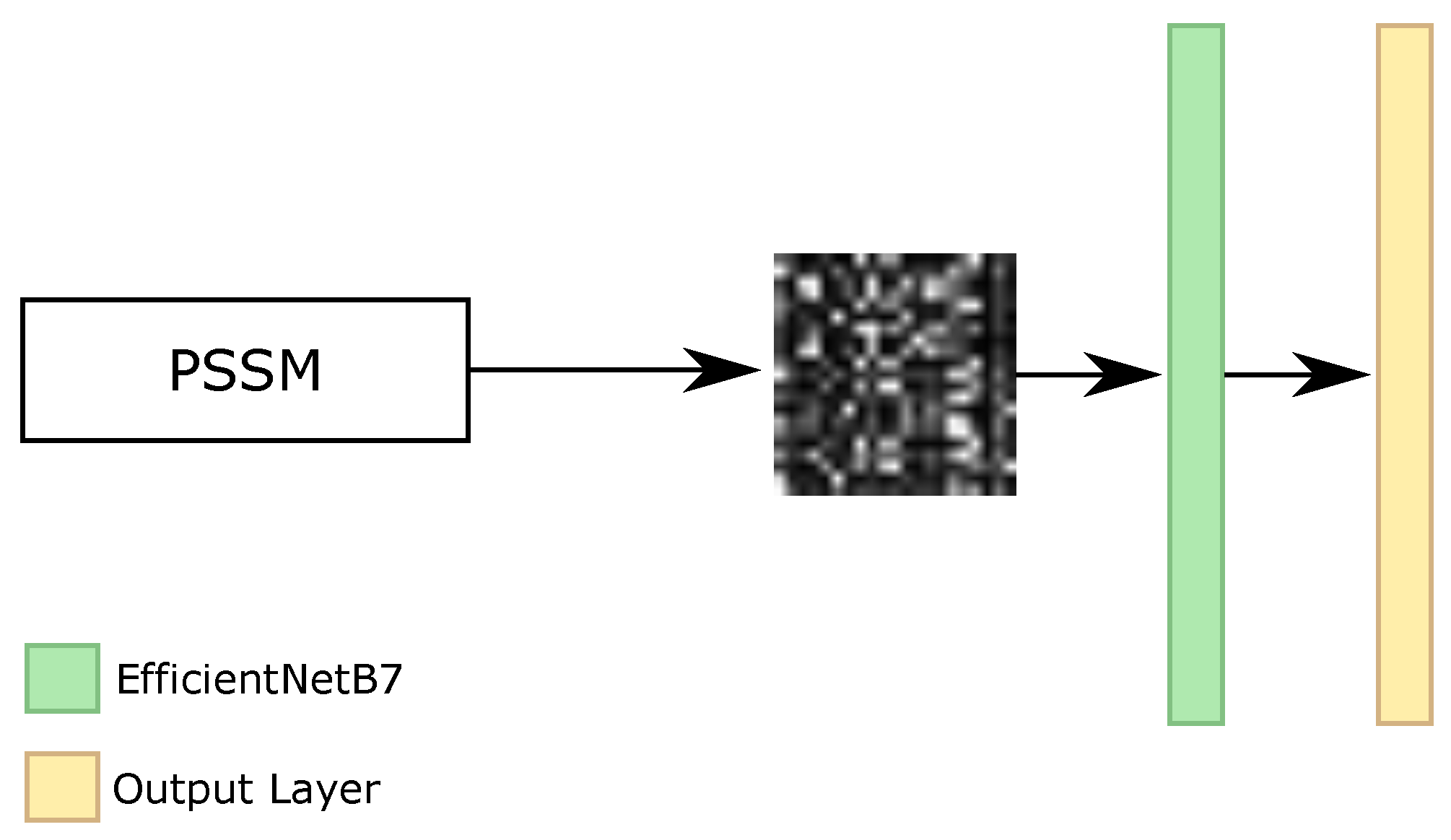
As our RNN, RF, Iv4B, and IRN classifiers use all of the features from the datasets, i.e., the amino acid sequence and PSSM information, and our BERT classifier only employed the amino acid sequence, we investigated one different classifier that applied only the PSSM features. Considering that this information only has values between 0 and 1, and each amino acid has 21 features for PSSM, the original format available in the datasets is the matrix of , where L represents the size of the sequence. With that, we analyzed the transformation of these characteristics as an image and the usage of convolutional neural networks (CNNs) for the image classification task.
With the image representation, we investigated the sliding window classification, i.e., we broke the sequence into parts and we made the classification of the secondary structure for the central amino acid. For the beginning and end of the proteins, we padded with values equal to 0. We evaluated different CNN architectures, such as EfficientNets [
67], ResNets [
68], and DenseNets [
69], as well as the size of the sliding window. The bests results achieved on the validation set were using EfficientNetB7 networks, with images with dimensions equal to 21 × 21, 63 × 63, 105 × 105, and 147 × 147. As the minimum size of input for the networks is 32 × 32, we resized the 21 × 21 image into 63 × 63. Each value of PSSM information was transformed to 3 × 3, 1 × 3, 1 × 5, and 1 × 7 pixels for 21 × 21, 63 × 63, 105 × 105, and 147 × 147 images, respectively.
Figure 9 illustrates the general architecture of CNN classifier.
We trained the EfficientNetB7 with TensorFlow [
56] framework for 50 epochs, with learning rate equal to
, categorical cross-entropy loss function, Adam [
59] optimizer, early stopping, and reducing learning rate by
after five epochs, without better results on the validation set.
Afterward, we made the ensemble between the four networks. The final ensemble between the EfficientNetB7 is considered the CNN classifier.
4.1.7. Ensemble of Template-Free Classifiers
After the predictions of RNN, RF, Iv4B, IRN, BERT, and CNN, we constructed the ensemble between them. We called this ensemble the template-free ensemble.
4.2. Template-Based Classifiers
Our template-based classifiers use BLAST to search for homologous proteins. We employed, as a searching database, all proteins with secondary structures determined by laboratory methods from PDB until 2018, and we used the retrieved proteins to make the predictions of the query’s secondary structures. As the protein query could be on the PDB database, we removed it from the retrieved results.
We created two different template-based classifiers. The first method, which we called specific alignments, considers only good alignments between the protein query and the homologies from PDB, while the second method, which we called general alignments, could retrieve general alignments, with fewer restrictions.
The execution required to search for homologous proteins (using BLAST) for specific alignments and general alignments classifiers, as well as for the ensemble of the template-based classifiers, is shown in
Table 23. For template-based classifiers, no training step is required, differently from template-free classifiers.
4.2.1. Specific Alignments
For the specific alignments classifier, we evaluated different restriction configurations of the retrieved homologies. We analyzed the number of alignments retrieved, the E-value configuration, and the use of weights to weight the retrieved proteins. We made a grid search with different parameters; the best results achieved on the validation set involved using the first 10 retrieved sequences, with decreasing weights, i.e., the first one received weights equal to 10, the second one received weights equal to 9, and so on, and the restriction of the E-value less than or equal to for the alignments. In the end, the probability of each structure was equal to the alignment voting.
As this classifier only considers good alignments, some amino acids can have no secondary structure predictions. In these cases, we consider that the probabilities of each class are equal to 0.
4.2.2. General Alignments
Our second template-based classifier, general alignments, is more general than the specific alignments classifiers. The general alignments classifier can predict all amino acid secondary structures.
We evaluated different parameters of this classifier, which included E-value configuration, the number of alignments retrieved, the use of local search, i.e., if the amino acid did not receive a prediction, we searched for neighbor amino acids that received a prediction. After the grid search, the best parameters found on the validation set was the E-value less than or equal to 10, using the top 100 retrieval sequences, with decreasing weights, i.e., the first one received weights equal to 100, the second one received weights equal to 99, and so on, and a local search of a size equal to 201, and if the window did not find a prediction, it could increase.
4.2.3. Ensemble of Template-Based Classifiers
After the prediction of the specific alignment and general alignment classifiers, we made the ensemble between them. We called this ensemble the template-based ensemble.
4.3. Ensemble of Template-Free and Template-Based Classifiers
After the ensemble of template-free methods and the ensemble of template-based classifiers, we made the ensemble between them, called the final ensemble. This process required 52 min for execution, considering the predictions on the CB6133 and CB513 datasets.
4.4. Ensemble Method
We used the method presented in this section to make the ensemble between each classifier, for instance, the ensemble of random forests, between classifiers of the same group, such as template-free classifiers, and the final ensemble between the template-free and template-based classifiers.
Our ensemble method uses the prediction of each classifier that will be fused, and it finds weights for each class of each classifier. For example, if we make the ensemble between five classifiers, the algorithm finds eight weights for each classifier, considering that there are eight possible classes, totaling forty weights. All of the weights were found using the predictions made on the validation set.
The core of our method is the genetic algorithm [
70]. The algorithm starts with 2000 individuals (representing the weights that will be associated with each class, for each classifier), with weights generated by uniform distribution, between 0 and 1. In each generation, we select the 100 best individuals by the highest Q8 accuracy to generate 900 new individual throw crossovers. These 1000 individuals (parents and new individuals) generate more 1000 individual throw mutations. At the end of each step, i.e., after crossovers and mutations, we normalize the individual dividing each weight by the maximum weight of this individual. We used 1000 generations and the early stopping technique, which stops the algorithm after 50 generations without better results.
Afterward, the algorithms select the top 100 individuals ordered by Q8 accuracy to make a local search. The top 100 individual generate 100 individual throw mutations. Again, we normalized each individual by dividing each weight by the maximum weight of this individual. We carry out this process by 1000 generations or if the early stopping stops the algorithm. In the end, the best individual, i.e., the best weights, based on Q8 accuracy, are used to make the ensemble between the classifiers.
4.5. Evaluation Metrics
We applied the Q8 accuracy, precision, and recall metrics to evaluate our method. Equation (
1) shows the Q8 accuracy, which is the most important metric used in the literature to compare different classifiers for the protein secondary structure prediction task.
Precision metric is presented in Equation (
2), where TP indicates the number of true positive and FP specifies the number of false positives. This metric evaluates the proportion of positive classifications of positive samples.
Equation (
3) shows the recall metric. In the equation, FN indicates the number of false negative samples. This metric evaluates the proportion of correctly classified positive data samples.
4.6. Datasets
In our study, we used two datasets, CB6133 and CB513. In this section, we present some relevant characteristics of these two datasets.
4.6.1. CB6133 Dataset
The CB6133 dataset is a set of 6133 proteins with sequences up to 700 amino acids, and each protein of this database has a maximum of 30% similarity between them [
7].
Each protein of the dataset has a matrix of 700 × 50 features, i.e., the first dimension of the matrix indicates the amino acids (proteins with less than 700 amino acids received padding). For each amino acid that forms the protein, there exist 50 features. The 21 first features indicate the letter that represents the amino acid in the one-hot encoding format (all 20 amino acids, for instance “A” for alanine, plus the special amino acid “X”), i.e., each amino acid only has one value equal to 1 and the remaining are equal to 0. The next 21 features were generated by the position-specific score matrix (PSSM) [
16]. The last 8 features are the 8 possible secondary structures in the one-hot encoding format. The padding amino acids have all the features equal to 0.
We used the same split of the dataset as applied in the literature; that is, 5600 proteins for training, 256 proteins for validation, and 272 proteins for testing. Thus, we are able to compare our results directly and fairly with the results in the literature.
Figure 10 presents the distribution of the secondary structures on the training, validation, and test sets, showing that the distributions on these three groups are similar. On the training and validation sets, there are structures of class “I”, while in the test set, there is no data of this class.
For the experiments involving the CB513 dataset, we employed a filtered version of the CB6133 for training and validation. This filtered version of the CB6133 only has proteins with less than 25% of similarity with the proteins of the CB513 dataset. We used the same split of this dataset as applied in the literature, i.e., 5278 proteins for training and 256 proteins for validation.
4.6.2. CB513 Dataset
The CB513 dataset is a set of 513 proteins that were used only in the test [
20]. Only one of the 513 proteins has more than 700 amino acids, so we decided to split this unique protein into two proteins, with the first one with 700 amino acids and the second one with the remaining amino acids.
As with the CB6133, all proteins of the CB513 dataset have 50 features, with the first 21 features related to the letter that represents the amino acid in the one-hot encoding format, the next 21 features indicate the PSSM information, and the remaining is related to the secondary structure in the one-hot encoding format. Again, proteins with less than 700 amino acids received padding with all values equal to 0.
Figure 11 illustrates the training, validation, and test set distributions of the secondary structures. The training and validation sets employed are from the filtered version of the CB6133 dataset, which has less than 25% of similarity to the proteins on the CB513 dataset. It shows a similar split to the CB6133 dataset. In the CB513 experiments, there exist data of class “I” in the training, validation, and test sets.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}