
Tau Biomarkers in Dementia: Positron Emission Tomography Radiopharmaceuticals in Tauopathy Assessment and Future Perspective

 ,
,  ,
,  ,
,
Abstract
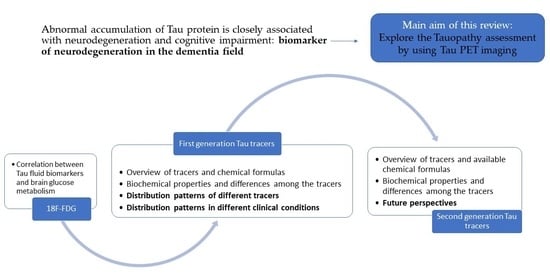
:
1. Introduction
1.1. Epidemiology
1.2. Neuropathology
1.3. Tau Deposition
1.4. Biomarkers in Dementia
1.5. PET Imaging in Dementia
1.6. Purpose of This Manuscript
2. Tauopathy and Brain Glucose Metabolism
3. Tau PET Imaging
3.1. First-Generation Tracers
3.1.1. [18F]FDDNP (C18H16FN3)
3.1.2. [11C]PBB3 (C17H15N3OS)
3.1.3. [18F]T808 (C17H19FN4)
3.1.4. THK Arylquinoline Series
3.1.5. [18F]Flortaucipir (Tauvid) (C16H10FN3)
3.2. Tau PET Tracers’ Distribution Pattern
3.2.1. Distribution Pattern of [18F]Flortaucipir
- —
- Pattern I (negative scan): absence of [18F]Flortaucipir signal in any brain area;
- —
- —
- —
- Pattern IV (non-AD-like): atypical [18F]FTP signal distribution, not following the expected Braak distribution of Tau tangles (for example, predominant white matter or subcortical binding consistent with non-AD syndromes) [107].
- —
- Patients with an amnestic-predominant presentation (n = 5) showed the highest [18F]Flortaucipir retention in medial temporal and lateral temporoparietal areas;
- —
- Patients with lvPPA (n = 5) demonstrated asymmetric left- greater than right-hemisphere [18F]Flortaucipir uptake in three of five patients;
- —
- In most AD patients who underwent all three PET exams, there was a strong negative association between [18F]Flortaucipir and [18F]FDG uptake and less pronounced positive associations between [11C]PiB and [18F]FDG and [18F]Flortaucipir and [11C]PiB;
- —
- In all patients, younger age was associated with greater [18F]Flortaucipir uptake in wide regions of the neocortex, while older age was associated with increased [18F]Flortaucipir in the medial temporal lobe.
3.2.2. Distribution Pattern of Other Tau Tracers
3.3. Second-Generation Tracers and Future Perspective
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jellinger, K.A. Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener. Dis. 2008, 5, 118–121. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, M.; Cimini, A.; Chiaravalloti, A.; Filippi, L.; Schillaci, O. Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia. Int. J. Mol. Sci. 2020, 21, 7481. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Schmitz, M.; Ferrer, I.; Zerr, I. CSF biomarkers in neurodegenerative and vascular dementias. Prog. Neurobiol. 2016, 138–140, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544, Erratum in 2017, 389, e1. [Google Scholar]
- World Health Organization. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Liu, Y.; Tan, L.; Wang, H.F.; Liu, Y.; Hao, X.K.; Tan, C.C.; Jiang, T.; Liu, B.; Zhang, D.Q.; Yu, J.T. Alzheimer’s Disease Neuroimaging Initiative. Multiple Effect of APOE Genotype on Clinical and Neuroimaging Biomarkers Across Alzheimer’s Disease Spectrum. Mol. Neurobiol. 2016, 53, 4539–4547. [Google Scholar] [CrossRef]
- Ricci, M.; Chiaravalloti, A.; Martorana, A.; Koch, G.; De Lucia, V.; Barbagallo, G.; Schillaci, O. The role of epsilon phenotype in brain glucose consumption in Alzheimer’s disease. Ann. Nucl. Med. 2020, 34, 254–262. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, 12–20. [Google Scholar] [CrossRef]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of Alzheimer’s disease—A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Ryan, N.S.; Rossor, M.N.; Fox, N.C. Alzheimer’s disease in the 100 years since Alzheimer’s death. Brain 2015, 138 Pt 12, 3816–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Singh, A. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Capetillo-Zarate, E.; Del Tredici, K.; Braak, H. The development of amyloid beta protein deposits in the aged brain. Sci. Aging Knowl. Environ. 2006, 2006, re1. [Google Scholar] [CrossRef]
- Reinhard, C.; Hébert, S.S.; De Strooper, B. The amyloid-beta precursor protein: Integrating structure with biological function. EMBO J. 2005, 24, 3996–4006. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. Beta-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging. 1997, 18, 351–357. [Google Scholar] [CrossRef]
- Sato, C.; Barthélemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 98, 861–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915–924, Erratum in 2019, 76, 986. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer’s disease. Acta Neuropathol. 1985, 68, 325–332. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Welikovitch, L.A.; Do Carmo, S.; Maglóczky, Z.; Szocsics, P.; Lőke, J.; Freund, T.; Cuello, A.C. Evidence of intraneuronal Aβ accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol. 2018, 136, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef]
- Wolfsgruber, S.; Jessen, F.; Koppara, A.; Kleineidam, L.; Schmidtke, K.; Frölich, L.; Kurz, A.; Schulz, S.; Hampel, H.; Heuser, I.; et al. Subjective cognitive decline is related to CSF biomarkers of AD in patients with MCI. Neurology 2015, 84, 1261–1268. [Google Scholar] [CrossRef]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Visser, P.J.; Verhey, F.; Knol, D.L.; Scheltens, P.; Wahlund, L.O.; Freund-Levi, Y.; Tsolaki, M.; Minthon, L.; Wallin, A.K.; Hampel, H.; et al. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: A prospective cohort study. Lancet Neurol. 2009, 8, 619–627. [Google Scholar] [CrossRef]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Londos, E.; Blennow, K.; Minthon, L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol. 2006, 5, 228–234, Erratum in 2006, 5, 293. [Google Scholar] [CrossRef] [Green Version]
- Wallin, A.K.; Blennow, K.; Zetterberg, H.; Londos, E.; Minthon, L.; Hansson, O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology 2010, 74, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Isaac, M.; Vamvakas, S.; Abadie, E.; Jonsson, B.; Gispen, C.; Pani, L. Qualification opinion of novel methodologies in the predementia stage of Alzheimer’s disease: Cerebro-spinal-fluid related biomarkers for drugs affecting amyloid burden—Regulatory considerations by European Medicines Agency focusing in improving benefit/risk in regulatory trials. Eur. Neuropsychopharmacol. 2011, 21, 781–788. [Google Scholar] [PubMed]
- Zetterberg, H.; Tullhög, K.; Hansson, O.; Minthon, L.; Londos, E.; Blennow, K. Low incidence of post-lumbar puncture headache in 1,089 consecutive memory clinic patients. Eur. Neurol. 2010, 63, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P. Alzheimer’s Disease Neuroimaging Initiative, Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O.; Swedish BioFINDER Study Group. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef]
- Reiman, E.M.; Jagust, W.J. Brain imaging in the study of Alzheimer’s disease. Neuroimage 2012, 61, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Lameka, K.; Farwell, M.D.; Ichise, M. Positron Emission Tomography. Handb. Clin. Neurol. 2016, 135, 209–227. [Google Scholar] [PubMed] [Green Version]
- Phelps, M.E.; Huang, S.C.; Hoffman, E.J.; Selin, C.; Sokoloff, L.; Kuhl, D.E. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: Validation of method. Ann. Neurol. 1979, 6, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Parent, M.J.; Souza, D.G.; Leuzy, A.; Lecrux, C.; Kim, H.I.; Gauthier, S.; Pellerin, L.; Hamel, E.; Rosa-Neto, P. [18F]FDG PET signal is driven by astroglial glutamate transport. Nat. Neurosci. 2017, 20, 393–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisoni, G.B.; Boccardi, M.; Barkhof, F.; Blennow, K.; Cappa, S.; Chiotis, K.; Démonet, J.F.; Garibotto, V.; Giannakopoulos, P.; Gietl, A.; et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017, 16, 661–676. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2009, 36, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, G.E.; Chen, K.; Pietrini, P.; Rapoport, S.I.; Reiman, E.M. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am. J. Psychiatry 2002, 159, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, S.; Frey, K.A.; Koeppe, R.A.; Foster, N.L.; Kuhl, D.E. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J. Nucl. Med. 1995, 36, 1238–1248. [Google Scholar] [PubMed]
- Herholz, K.; Carter, S.F.; Jones, M. Positron emission tomography imaging in dementia. Br. J. Radiol. 2007, 80, S160–S167. [Google Scholar] [CrossRef] [PubMed]
- Rubí, S.; Noguera, A.; Tarongí, S.; Oporto, M.; García, A.; Vico, H.; Espino, A.; Picado, M.J.; Mas, A.; Peña, C.; et al. Concordance between brain 18F-FDG PET and cerebrospinal fluid biomarkers in diagnosing Alzheimer’s disease. Rev. Esp. Med. Nucl. Imagen Mol. (Engl. Ed.) 2018, 37, 3–8. [Google Scholar] [PubMed]
- Yakushev, I.; Muller, M.J.; Buchholz, H.G.; Lang, U.; Rossmann, H.; Hampel, H.; Schreckenberger, M.; Fellgiebel, A. Stage-dependent agreement between cerebrospinal fluid proteins and FDG-PET findings in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 241–247. [Google Scholar] [CrossRef]
- Santangelo, R.; Masserini, F.; Agosta, F.; Sala, A.; Caminiti, S.P.; Cecchetti, G.; Caso, F.; Martinelli, V.; Pinto, P.; Passerini, G.; et al. CSF p-tau/Aβ42 ratio and brain FDG-PET may reliably detect MCI "imminent" converters to AD. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalloti, A.; Martorana, A.; Koch, G.; Toniolo, S.; di Biagio, D.; di Pietro, B.; Schillaci, O. Functional correlates of t-Tau, p-Tau and Aβ1−42 amyloid cerebrospinal fluid levels in Alzheimer’s disease: A ¹⁸F-FDG PET/CT study. Nucl. Med. Commun. 2015, 36, 461–468. [Google Scholar] [CrossRef]
- Chiaravalloti, A.; Barbagallo, G.; Ricci, M.; Martorana, A.; Ursini, F.; Sannino, P.; Karalis, G.; Schillaci, O. Brain metabolic correlates of CSF Tau protein in a large cohort of Alzheimer’s disease patients: A CSF and FDG PET study. Brain Res. 2018, 1678, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Jaillard, A.; Vanhoutte, M.; Maureille, A.; Schraen, S.; Skrobala, E.; Delbeuck, X.; Rollin-Sillaire, A.; Pasquier, F.; Bombois, S.; Semah, F. The relationship between CSF biomarkers and cerebral metabolism in early-onset Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalloti, A.; Barbagallo, G.; Martorana, A.; Castellano, A.E.; Ursini, F.; Schillaci, O. Brain metabolic patterns in patients with suspected non-Alzheimer’s pathophysiology (SNAP) and Alzheimer’s disease (AD): Is [18F] FDG a specific biomarker in these patients? Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1796–1805. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Chételat, G.; Dickson, D.; Fagan, A.M.; Frisoni, G.B.; Jagust, W.; Mormino, E.C.; Petersen, R.C.; Sperling, R.A.; et al. Suspected non-Alzheimer disease pathophysiology—Concept and controversy. Nat. Rev. Neurol. 2016, 12, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, C.; Chiaravalloti, A.; Nuccetelli, M.; Izzi, F.; Sancesario, G.; Cimini, A.; Bernardini, S.; Schillaci, O.; Mercuri, N.B.; Fabio, P. Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer’s disease. J. Neurol. 2017, 264, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Baghel, V.; Tripathi, M.; Parida, G.; Gupta, R.; Yadav, S.; Kumar, P.; Dey, A.B.; Damle, N.A.; Kumar, R.; Bal, C. In Vivo Assessment of Tau Deposition in Alzheimer Disease and Assessing Its Relationship to Regional Brain Glucose Metabolism and Cognition. Clin. Nucl. Med. 2019, 44, e597–e601. [Google Scholar] [CrossRef] [PubMed]
- Nobili, F.; Arbizu, J.; Bouwman, F.; Drzezga, A.; Agosta, F.; Nestor, P.; Walker, Z.; Boccardi, M. EANM-EAN Task Force for the Prescription of FDG-PET for Dementing Neurodegenerative Disorders. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain 18 F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur. J. Neurol. 2018, 25, 1201–1217. [Google Scholar] [PubMed] [Green Version]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, P.F.; Pichet Binette, A.; Gonneaud, J.; Breitner, J.C.S.; Villeneuve, S. Characterization of Alzheimer Disease Biomarker Discrepancies Using Cerebrospinal Fluid Phosphorylated Tau and AV1451 Positron Emission Tomography. JAMA Neurol. 2020, 77, 508–516, Erratum in 2021, 78, 370. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Fumagalli, M.A.; Shrivastava, P.; Aguilar-Pineda, J.A.; Nieto-Montesinos, R.; Carpio, D.D.; Peralta-Mestas, A.; Caracela-Zeballos, C.; Valdez-Lazo, G.; Fernandez-Macedo, V.; Pino-Figueroa, A.; et al. Diagnosis of Alzheimer’s Disease in Developed and Developing Countries: Systematic Review and Meta-Analysis of Diagnostic Test Accuracy. J. Alzheimer’s Dis. Rep. 2021, 5, 15–30. [Google Scholar] [CrossRef]
- Smith, A.; Puschmann, A.; Schöll, M.; Ohlsson, T.; van Swieten, J.; Honer, M.; Englund, E.; Hansson, O. 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016, 139, 2372–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Knopman, D.S.; Graff-Radford, J.; Syrjanen, J.A.; Senjem, M.L.; Schwarz, C.G.; Dheel, C.; Wszolek, Z.; Rademakers, R.; Kantarci, K.; et al. In vivo 18F-AV-1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology 2018, 90, e947–e954. [Google Scholar] [CrossRef] [PubMed]
- La Joie, R.; Visani, A.V.; Lesman-Segev, O.H.; Baker, S.L.; Edwards, L.; Iaccarino, L.; Soleimani-Meigooni, D.N.; Mellinger, T.; Janabi, M.; Miller, Z.A.; et al. Association of APOE4 and Clinical Variability in Alzheimer Disease With the Pattern of Tau- and Amyloid-PET. Neurology 2021, 96, e650–e661. [Google Scholar] [CrossRef]
- Harada, R.; Okamura, N.; Furumoto, S.; Tago, T.; Yanai, K.; Arai, H.; Kudo, Y. Characteristics of Tau and Its Ligands in PET Imaging. Biomolecules 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.W.; Goto, T.K.; Leung, W.K. The Changing Landscape of Neuroscience Research, 2006–2015: A Bibliometric Study. Front. Neurosci. 2017, 11, 120. [Google Scholar] [CrossRef]
- Wang, Y.T.; Edison, P. Tau Imaging in Neurodegenerative Diseases Using Positron Emission Tomography. Curr. Neurol. Neurosci. Rep. 2019, 19, 45. [Google Scholar] [CrossRef] [Green Version]
- Jie, C.V.M.L.; Treyer, V.; Schibli, R.; Mu, L. Tauvid™: The First FDA-Approved PET Tracer for Imaging Tau Pathology in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 110. [Google Scholar] [CrossRef]
- Shin, J.; Kepe, V.; Barrio, J.R.; Small, G.W. The merits of FDDNP-PET imaging in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, G.W.; Agdeppa, E.D.; Kepe, V.; Satyamurthy, N.; Huang, S.C.; Barrio, J.R. In vivo brain imaging of tangle burden in humans. J. Mol. Neurosci. 2002, 19, 323–327. [Google Scholar] [CrossRef]
- Boxer, A.L.; Rabinovici, G.D.; Kepe, V.; Goldman, J.; Furst, A.J.; Huang, S.C.; Baker, S.L.; O’neil, J.P.; Chui, H.; Geschwind, M.D.; et al. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology 2007, 69, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.D.; Siddarth, P.; Kepe, V.; Scheibel, K.E.; Huang, S.C.; Barrio, J.R.; Small, G.W. Positron emission tomography of brain β-amyloid and τ levels in adults with Down syndrome. Arch. Neurol. 2011, 68, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.R.; Trojanowski, J.Q.; Lee, V.M.Y.; Ono, M.; et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, N.; Harada, R.; Furumoto, S.; Arai, H.; Yanai, K.; Kudo, Y. Tau PET imaging in Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2014, 14, 500. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Kawamura, K.; Igarashi, N.; Takei, M.; Fujishiro, T.; Aihara, Y.; Shiomi, S.; Muto, M.; Ito, T.; Furutsuka, K.; et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J. Nucl. Med. 2014, 55, 1532–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahata, K.; Kimura, Y.; Sahara, N.; Koga, S.; Shimada, H.; Ichise, M.; Saito, F.; Moriguchi, S.; Kitamura, S.; Kubota, M.; et al. PET-detectable tau pathology correlates with long-term neuropsychiatric outcomes in patients with traumatic brain injury. Brain 2019, 142, 3265–3279. [Google Scholar] [CrossRef]
- Ono, M.; Sahara, N.; Kumata, K.; Ji, B.; Ni, R.; Koga, S.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.; Yoshida, M.; et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017, 140, 764–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, D.T.; Szardenings, A.K.; Bahri, S.; Walsh, J.C.; Mu, F.; Xia, C.; Shankle, W.R.; Lerner, A.J.; Su, M.Y.; Elizarov, A.; et al. Early clinical PET imaging results with the novel PHF-tau radioligand [18F]-T808. J. Alzheimers Dis. 2014, 38, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, N.; Suemoto, T.; Furumoto, S.; Suzuki, M.; Shimadzu, H.; Akatsu, H.; Yamamoto, T.; Fujiwara, H.; Nemoto, M.; Maruyama, M.; et al. Quinoline and benzimidazole derivatives: Candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J. Neurosci. 2005, 25, 10857–10862. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Furumoto, S.; Harada, R.; Tago, T.; Yoshikawa, T.; Fodero-Tavoletti, M.; Mulligan, R.S.; Villemagne, V.L.; Akatsu, H.; Yamamoto, T.; et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J. Nucl. Med. 2013, 54, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Murugan, N.A.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Ågren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: Evidence from in silico modelling and in vivo imaging. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.P.; Pascoal, T.A.; Mathotaarachchi, S.; Therriault, J.; Kang, M.S.; Shin, M.; Guiot, M.C.; Guo, Q.; Harada, R.; Comley, R.A.; et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimer’s Res Ther. 2017, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, R.; Ishiki, A.; Kai, H.; Sato, N.; Furukawa, K.; Furumoto, S.; Tago, T.; Tomita, N.; Watanuki, S.; Hiraoka, K.; et al. Correlations of 18F-THK5351 PET with Postmortem Burden of Tau and Astrogliosis in Alzheimer Disease. J. Nucl. Med. 2018, 59, 671–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Makaretz, S.J.; Caso, C.; McGinnis, S.; Gomperts, S.N.; Sepulcre, J.; Gomez-Isla, T.; Hyman, B.T.; Schultz, A.; Vasdev, N.; et al. Association of In Vivo [18F]AV-1451 Tau PET Imaging Results With Cortical Atrophy and Symptoms in Typical and Atypical Alzheimer Disease. JAMA Neurol. 2017, 74, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S.; Pontecorvo, M.J.; Devous, M.D.; Lu, M.; Arora, A.K.; Truocchio, S.P.; Aldea, P.; Flitter, M.; Locascio, T.; Devine, M.; et al. Positron Emission Tomography Imaging with [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes. JAMA Neurol. 2020, 77, 829–839. [Google Scholar] [CrossRef] [PubMed]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Schonhaut, D.R.; McMillan, C.T.; Spina, S.; Dickerson, B.C.; Siderowf, A.; Devous, M.D.; Tsai, R.; Winer, J.; Russell, D.S.; Litvan, I.; et al. 18F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: A multicenter study. Ann. Neurol. 2017, 82, 622–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeiren, C.; Motte, P.; Viot, D.; Mairet-Coello, G.; Courade, J.P.; Citron, M.; Mercier, J.; Hannestad, J.; Gillard, M. The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases. Mov. Disord. 2017, 33, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Przybelski, S.A.; Lesnick, T.G.; Lemstra, A.W.; Londos, E.; Blanc, F.; Nedelska, Z.; Schwarz, C.G.; Graff-Radford, J.; Senjem, M.L.; et al. β-Amyloid and tau biomarkers and clinical phenotype in dementia with Lewy bodies. Neurology 2020, 95, e3257–e3268. [Google Scholar] [CrossRef]
- Tudorascu, D.L.; Laymon, C.M.; Zammit, M.; Minhas, D.S.; Anderson, S.J.; Ellison, P.A.; Zaman, S.; Ances, B.M.; Sabbagh, M.; Johnson, S.C.; et al. Relationship of amyloid beta and neurofibrillary tau deposition in Neurodegeneration in Aging Down Syndrome (NiAD) study at baseline. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, 1–8. [Google Scholar]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.W.; Mattsson, N.; Iturria-Medina, Y.; Strandberg, O.T.; Schöll, M.; Dansereau, C.; Villeneuve, S.; van der Flier, W.M.; Scheltens, P.; Bellec, P.; et al. Alzheimer’s Disease Neuroimaging Initiative; Swedish BioFINDER Study. Data-driven approaches for tau-PET imaging biomarkers in Alzheimer’s disease. Hum. Brain Mapp. 2019, 40, 638–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golla, S.S.; Wolters, E.E.; Timmers, T.; Ossenkoppele, R.; van der Weijden, C.W.; Scheltens, P.; Schwarte, L.; Mintun, M.A.; Devous, M.D.S.; Schuit, R.C.; et al. Parametric methods for [18F]flortaucipir PET. J. Cereb. Blood Flow Metab. 2020, 40, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barret, O.; Alagille, D.; Sanabria, S.; Comley, R.A.; Weimer, R.M.; Borroni, E.; Mintun, M.; Seneca, N.; Papin, C.; Morley, T.; et al. Kinetic Modeling of the Tau PET Tracer 18F-AV-1451 in Human Healthy Volunteers and Alzheimer Disease Subjects. J. Nucl. Med. 2017, 58, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, T.; Ossenkoppele, R.; Wolters, E.E.; Verfaillie, S.C.J.; Visser, D.; Golla, S.S.V.; Barkhof, F.; Scheltens, P.; Boellaard, R.; van der Flier, W.M.; et al. Associations between quantitative [18F]flortaucipir tau PET and atrophy across the Alzheimer’s disease spectrum. Alzheimer’s Res. Ther. 2019, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Lowe, V.J.; Wiste, H.J.; Senjem, M.L.; Weigand, S.D.; Therneau, T.M.; Boeve, B.F.; Josephs, K.A.; Fang, P.; Pandey, M.K.; Murray, M.E.; et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 2018, 141, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Sonni, I.; Lesman Segev, O.H.; Baker, S.L.; Iaccarino, L.; Korman, D.; Rabinovici, G.D.; Jagust, W.J.; Landau, S.M.; La Joie, R.; Alzheimer’s Disease Neuroimaging Initiative. Evaluation of a visual interpretation method for tau-PET with 18F-flortaucipir. Alzheimer’s Dement. 2020, 12, e12133. [Google Scholar]
- Tetzloff, K.A.; Graff-Radford, J.; Martin, P.R.; Tosakulwong, N.; Machulda, M.M.; Duffy, J.R.; Clark, H.M.; Senjem, M.L.; Schwarz, C.G.; Spychalla, A.J.; et al. Regional Distribution, Asymmetry, and Clinical Correlates of Tau Uptake on [18F]AV-1451 PET in Atypical Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1713–1724. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Schonhaut, D.R.; Schöll, M.; Lockhart, S.N.; Ayakta, N.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Lazaris, A.; Cantwell, A.; et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016, 139, 1551–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitwell, J.L. Multimodal neuroimaging provides insights into the biology of Alzheimer’s disease. Brain 2018, 141, 326–329. [Google Scholar] [CrossRef]
- Hansson, O.; Grothe, M.J.; Strandberg, T.O.; Ohlsson, T.; Hägerström, D.; Jögi, J.; Smith, R.; Schöll, M. Tau Pathology Distribution in Alzheimer’s disease Corresponds Differentially to Cognition-Relevant Functional Brain Networks. Front. Neurosci. 2017, 31, 11–167. [Google Scholar] [CrossRef] [PubMed]
- Yap, S.Y.; Frias, B.; Wren, M.C.; Schöll, M.; Fox, N.C.; Årstad, E.; Lashley, T.; Sander, K. Discriminatory ability of next-generation tau PET tracers for Alzheimer’s disease. Brain 2021, 144, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Okamura, N.; Furukawa, K.; Furumoto, S.; Harada, R.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Ishikawa, Y.; Tago, T.; et al. Longitudinal Assessment of Tau Pathology in Patients with Alzheimer’s Disease Using [18F]THK-5117 Positron Emission Tomography. PLoS ONE 2015, 10, e0140311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, R.; Okamura, N.; Furumoto, S.; Furukawa, K.; Ishiki, A.; Tomita, N.; Tago, T.; Hiraoka, K.; Watanuki, S.; Shidahara, M.; et al. 18F-THK5351: A Novel PET Radiotracer for Imaging Neurofibrillary Pathology in Alzheimer Disease. J. Nucl. Med. 2016, 57, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tsui, W.; Rusinek, H.; Butler, T.; Mosconi, L.; Pirraglia, E.; Mozley, D.; Vallabhajosula, S.; Harada, R.; Furumoto, S.; et al. Cortical laminar binding of PET amyloid and tau tracers in Alzheimer disease. J. Nucl. Med. 2015, 56, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiotis, K.; Stenkrona, P.; Almkvist, O.; Stepanov, V.; Ferreira, D.; Arakawa, R.; Takano, A.; Westman, E.; Varrone, A.; Okamura, N.; et al. Dual tracer tau PET imaging reveals different molecular targets for 11C-THK5351 and 11C-PBB3 in the Alzheimer brain. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiotis, K.; Saint-Aubert, L.; Rodriguez-Vieitez, E.; Leuzy, A.; Almkvist, O.; Savitcheva, I.; Jonasson, M.; Lubberink, M.; Wall, A.; Antoni, G.; et al. Longitudinal changes of tau PET imaging in relation to hypometabolism in prodromal and Alzheimer’s disease dementia. Mol. Psychiatry 2018, 23, 1666–1673. [Google Scholar] [CrossRef]
- Endo, H.; Shimada, H.; Sahara, N.; Ono, M.; Koga, S.; Kitamura, S.; Niwa, F.; Hirano, S.; Kimura, Y.; Ichise, M.; et al. In vivo binding of a tau imaging probe, [11C]PBB3, in patients with progressive supranuclear palsy. Mov. Disord. 2019, 34, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Wren, M.C.; Lashley, T.; Årstad, E.; Sander, K. Large inter- and intra-case variability of first generation tau PET ligand binding in neurodegenerative dementias. Acta Neuropathol. Commun. 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Schöll, M.; Leuzy, A.; Jögi, J.; Ohlsson, T.; Strandberg, O.; Hansson, O. Head-to-head comparison of tau positron emission tomography tracers [18F]flortaucipir and [18F]RO948. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.E.; Janssens, L.; Moechars, D.; Rombouts, F.J.R.; Timmers, M.; Barret, O.; Constantinescu, C.C.; Madonia, J.; Russell, D.S.; Sandiego, C.M.; et al. Clinical evaluation of [18F] JNJ-64326067, a novel candidate PET tracer for the detection of tau pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Therriault, J.; Benedet, A.L.; Savard, M.; Lussier, F.Z.; Chamoun, M.; Tissot, C.; Qureshi, M.N.I.; Kang, M.S.; Mathotaarachchi, S.; et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 2020, 143, 2818–2830. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Benedet, A.L.; Tudorascu, D.L.; Therriault, J.; Mathotaarachchi, S.; Savard, M.; Lussier, F.Z.; Tissot, C.; Chamoun, M.; Kang, M.S.; et al. Longitudinal 18F-MK-6240 tau tangles accumulation follows Braak stages. Brain 2021, 13, awab248. [Google Scholar] [CrossRef]
- Lohith, T.G.; Bennacef, I.; Vandenberghe, R.; Salinas, C.A.; Declercq, R.; Reynders, T.; Telan-Choing, N.F.; Riffel, K.; Celen, S.; Serdons, K. Brain Imaging of Alzheimer Dementia Patients and Elderly Controls with 18F-MK-6240, a PET Tracer Targeting Neurofibrillary Tangles. J. Nucl. Med. 2019, 60, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Salinas, C.; Lohith, T.G.; Purohit, A.; Struyk, A.; Sur, C.; Bennacef, I.; Beaver, J.; Martarello, L. Test-retest characteristic of [18F]MK-6240 quantitative outcomes in cognitively normal adults and subjects with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2020, 40, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Krishnadas, N.; Doré, V.; Lamb, F.; Groot, C.; McCrory, P.; Guzman, R.; Mulligan, R.; Huang, K.; O’Donnell, M.; Ponsford, J.; et al. Case Report: 18F-MK6240 Tau Positron Emission Tomography Pattern Resembling Chronic Traumatic Encephalopathy in a Retired Australian Rules Football Player. Front. Neurol. 2020, 11, 598980. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Fu, J.; Yu, J.; Zhao, Q.; Guan, Y.; Zuo, C.; Li, M.; Tan, H.; Cheng, X. Tau PET Imaging with [18F]PM-PBB3 in Frontotemporal Dementia with MAPT Mutation. J. Alzheimer’s Dis. 2020, 76, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Tagai, K.; Ono, M.; Kubota, M.; Kitamura, S.; Takahata, K.; Seki, C.; Takado, Y.; Shinotoh, H.; Sano, Y.; Yamamoto, Y.; et al. High-Contrast In Vivo Imaging of Tau Pathologies in Alzheimer’s and Non-Alzheimer’s Disease Tauopathies. Neuron 2021, 109, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Bullich, S.; Barret, O.; Madonia, J.; Berndt, M.; Papin, C.; Perrotin, A.; Koglin, N.; Kroth, H.; Pfeifer, A.; et al. Tau PET imaging with 18F-PI-2620 in Patients with Alzheimer Disease and Healthy Controls: A First-in-Humans Study. J. Nucl. Med. 2020, 61, 911–919. [Google Scholar] [CrossRef]
- Brendel, M.; Barthel, H.; van Eimeren, T.; Marek, K.; Beyer, L.; Song, M.; Palleis, C.; Gehmeyr, M.; Fietzek, U.; Respondek, G.; et al. Assessment of 18F-PI-2620 as a Biomarker in Progressive Supranuclear Palsy. JAMA Neurol. 2020, 77, 1408–1419. [Google Scholar] [CrossRef]
- Beyer, L.; Nitschmann, A.; Barthel, H.; van Eimeren, T.; Unterrainer, M.; Sauerbeck, J.; Marek, K.; Song, M.; Palleis, C.; Respondek, G.; et al. Early-phase [18F]PI-2620 tau-PET imaging as a surrogate marker of neuronal injury. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2911–2922. [Google Scholar] [CrossRef] [PubMed]
- Frantellizzi, V.; Pani, A.; Ricci, M.; Locuratolo, N.; Fattapposta, F.; De Vincentis, G. Neuroimaging in Vascular Cognitive Impairment and Dementia: A Systematic Review. J. Alzheimer’s Dis. 2020, 73, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Perani, D. Brain Molecular Connectivity in Neurodegenerative Diseases: Recent Advances and New Perspectives Using Positron Emission Tomography. Front. Neurosci. 2019, 13, 617. [Google Scholar] [CrossRef] [Green Version]
- Chiaravalloti, A.; Fuccillo, E.; Martorana, A.; Ricci, M.; Giacomini, P.G.; Schillaci, O.; Di Girolamo, S. Hearing and cognitive impairment: A functional evaluation of associative brain areas in patients affected by Alzheimer’s disease. Funct. Neurol. 2019, 34, 15–20. [Google Scholar] [PubMed]
- Yang, F.; Roy Chowdhury, S.; Jacobs, H.I.L.; Johnson, K.A.; Dutta, J. A longitudinal model for tau aggregation in Alzheimer’s disease based on structural connectivity. Inf. Process. Med. Imaging 2019, 11492, 384–393. [Google Scholar]
- Franzmeier, N.; Dewenter, A.; Frontzkowski, L.; Dichgans, M.; Rubinski, A.; Neitzel, J.; Smith, R.; Strandberg, O.; Ossenkoppele, R.; Buerger, K.; et al. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease. Sci. Adv. 2020, 6, eabd1327. [Google Scholar] [CrossRef] [PubMed]
- Franzmeier, N.; Neitzel, J.; Rubinski, A.; Smith, R.; Strandberg, O.; Ossenkoppele, R.; Hansson, O.; Ewers, M.; Alzheimer’s Disease Neuroimaging Initiative (ADNI). Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat. Commun. 2020, 11, 347. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Tau Tracer | Chemical Formulas | ClinicalTrials.gov Identifier | Title | Condition or Disease |
|---|---|---|---|---|
| [18F]RO6958948 | NCT03174938 | The Swedish BioFINDER 2 Study | Dementia, AD, PD, Lewy body disease, etc. | |
| NCT02792179 | Evaluation of [18F] RO6958948 as Tracer for Positron Emission Tomography (PET) Imaging of Tau Burden in Alzheimer′s Disease Participants | AD | ||
| NCT02187627 | Evaluation of [11C]RO6924963, [11C]RO6931643, and [18F] RO6958948 as Tracers for Tau Imaging with Positron Emission Tomography in Healthy Control Subjects and Subjects with Alzheimer′s Disease | Healthy controls, AD | ||
| 11C-RO6924963; 11C-RO6931643 | NCT02187627 | Evaluation of [11C]RO6924963, [11C]RO6931643, and [18F] RO6958948 as Tracers for Tau Imaging with Positron Emission Tomography in Healthy Control Subjects and Subjects with Alzheimer′s Disease | Healthy controls, AD | |
| [18F]PM-PBB3 | C20H20FN3O2S | NCT04248270 | A Novel Tau Tracer in Young Onset Dementia | AD, Vascular dementia, dementia |
| NCT03625128 | [18F]PM-PBB3 PET Study in Tauopathy Including Alzheimer′s Disease, Other Dementias and Normal Controls | AD, CBS, Frontotemporal Dementia, PSP, etc. | ||
| NCT04169126 | Topography Staging and Dual Phase Image Quantification of Tau PET in Cognitive Impairment Subjects | AD | ||
| [18F MK-6240 | C16H11FN4 | NCT04104659 | Study of Tau Imaging with the Use of [18F]MK-6240 Tracer | AD |
| NCT03860857 | MRI and PET Biomarkers for Cognitive Decline in Older Adults | AD, Cognitive impairment and decline | ||
| NCT03706261 | Alzheimer′s PET Imaging in Racially/Ethnically Diverse Adults | AD | ||
| NCT04784416 | Transcranial Photobiomodulation for Alzheimer′s Disease (TRAP-AD) | MCI, AD | ||
| NCT03071224 | Phase 1 Evaluation of [18F]MK-6240 PET as an Imaging Marker for Tau Protein | AD, Healthy | ||
| NCT04437290 | University of Washington Alzheimer′s Disease Research Center (UW ADRC) Imaging & Biomarker Core | AD | ||
| [18F]PI2620 | C15H9FN4 | NCT04715750 | Evaluation of Imaging Characteristics of [18F]PI-2620 PET in AD and PSP Patients Using High and Low Specific Activity | AD, PSP |
| NCT04566003 | Evaluation Comparing Two Tau PET Radiotracers, [18F]PI-2620 and [18F]GTP1, in Subjects With Normal Condition or Prodromal to Moderate Alzheimer′s Disease | AD | ||
| [18F]GTP1 | NCT04566003 | Evaluation Comparing Two Tau PET Radiotracers, [18F]PI-2620 and [18F]GTP1, in Subjects With Normal Condition or Prodromal to Moderate Alzheimer′s Disease | AD | |
| NCT02640092 | Longitudinal Evaluation of [18F]GTP1 as a PET Radioligand for Imaging Tau in the Brain of Participants With Alzheimer′s Disease Compared to Healthy Participants | AD | ||
| [18F]MNI-958 | C20H20FN3O2S | NCT03545789 | Phase 1 Test-retest Evaluation of [18F]MNI-958 PET | Healthy, AD, PSP |
| NCT03058965 | Phase 0 Evaluation of [18F]MNI-958 as a Potential PET Radioligand for Imaging Tau Protein in the Brain | Healthy, AD, PSP | ||
| [18F]MNI-952 | NCT03080051 | Evaluation of [18F]MNI-952 as a Potential PET Radioligand for Imaging Tau Protein in the Brain | Healthy, AD, PSP | |
| [18F]MNI-1020 | NCT03239561 | Evaluation of Tau Protein in the Brain of Participants with Alzheimer′s Disease Compared to Healthy Participants | AD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, M.; Cimini, A.; Camedda, R.; Chiaravalloti, A.; Schillaci, O. Tau Biomarkers in Dementia: Positron Emission Tomography Radiopharmaceuticals in Tauopathy Assessment and Future Perspective. Int. J. Mol. Sci. 2021, 22, 13002. https://doi.org/10.3390/ijms222313002
Ricci M, Cimini A, Camedda R, Chiaravalloti A, Schillaci O. Tau Biomarkers in Dementia: Positron Emission Tomography Radiopharmaceuticals in Tauopathy Assessment and Future Perspective. International Journal of Molecular Sciences. 2021; 22(23):13002. https://doi.org/10.3390/ijms222313002
Chicago/Turabian StyleRicci, Maria, Andrea Cimini, Riccardo Camedda, Agostino Chiaravalloti, and Orazio Schillaci. 2021. "Tau Biomarkers in Dementia: Positron Emission Tomography Radiopharmaceuticals in Tauopathy Assessment and Future Perspective" International Journal of Molecular Sciences 22, no. 23: 13002. https://doi.org/10.3390/ijms222313002
APA StyleRicci, M., Cimini, A., Camedda, R., Chiaravalloti, A., & Schillaci, O. (2021). Tau Biomarkers in Dementia: Positron Emission Tomography Radiopharmaceuticals in Tauopathy Assessment and Future Perspective. International Journal of Molecular Sciences, 22(23), 13002. https://doi.org/10.3390/ijms222313002