
New Insights into the Pathogenesis of Systemic Mastocytosis

Department of Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Hannover Medical School, 30625 Hannover, Germany
Int. J. Mol. Sci. 2021, 22(9), 4900; https://doi.org/10.3390/ijms22094900
Submission received: 31 March 2021
/
Revised: 29 April 2021
/
Accepted: 30 April 2021
/
Published: 5 May 2021
(This article belongs to the Special Issue Mast Cells: From Host Defense to Pathology)
Abstract
:Mastocytosis is a type of myeloid neoplasm characterized by the clonal, neoplastic proliferation of morphologically and immunophenotypically abnormal mast cells that infiltrate one or more organ systems. Systemic mastocytosis (SM) is a more aggressive variant of mastocytosis with extracutaneous involvement, which might be associated with multi-organ dysfunction or failure and shortened survival. Over 80% of patients with SM carry the KIT D816V mutation. However, the KIT D816V mutation serves as a weak oncogene and appears to be a late event in the pathogenesis of mastocytosis. The management of SM is highly individualized and was largely palliative for patients without a targeted form of therapy in past decades. Targeted therapy with midostaurin, a multiple kinase inhibitor that inhibits KIT, has demonstrated efficacy in patients with advanced SM. This led to the recent approval of midostaurin by the United States Food and Drug Administration and European Medicines Agency. However, the overall survival of patients treated with midostaurin remains unsatisfactory. The identification of genetic and epigenetic alterations and understanding their interactions and the molecular mechanisms involved in mastocytosis is necessary to develop rationally targeted therapeutic strategies. This review briefly summarizes recent developments in the understanding of SM pathogenesis and potential treatment strategies for patients with SM.
1. Introduction
Mastocytosis is a myeloid neoplasm characterized by clonal expansion and the accumulation of morphologically and immunophenotypically abnormal mast cells (MCs) in one or more organ systems [1,2]. Mastocytosis patients show heterogeneous clinical presentations. The skin-limited presentation, cutaneous mastocytosis (CM), is particularly common among pediatric patients, with disease onset most frequently occurring within the first 2 years of life and commonly experiencing spontaneous regression of skin lesions, In contrast to CM, systemic mastocytosis (SM) associated with extracutaneous involvement is generally observed in adult patients. SM is generally more aggressive and may be associated with multi-organ dysfunction or failure and shortened survival [3,4,5,6]. The World Health Organization (WHO) classification of SM has been demonstrated to be prognostically relevant. The WHO classification is a useful first step for establishing prognosis, based on the following categories: indolent SM (ISM), aggressive SM (ASM), SM associated with a clonal hematological non-MC lineage disease (SM-AHNMD, now known as SM with an associated hematological neoplasm [SM-AHN] per the 2016 WHO classification guidelines [7]), and mast cell leukemia (MCL) [3,5]. Advanced SM (ASM, SM-AHN, and MCL) is generally associated with a poor prognosis, whereas ISM patients usually have a comparable life expectancy as the general population.
The molecular mechanisms that underlie SM development are not well-understood. Over 80% of patients with SM harbor the KIT D816V mutation [1]. However, KIT D816V mutation is a weak oncogene and appears to be a late event in the pathogenesis of mastocytosis [8]. Although KIT inhibitors present good inhibitory effects against MCs in vitro, treatment with KIT inhibitors alone has been disappointing in most published clinical trials for mastocytosis [1,4,9], likely due to the development of resistance against kinase inhibitors. The management of SM is typically highly individualized and was largely palliative for patients without a targeted form of therapy in past decades [10]. Recently, the US Food and Drug Administration (US FDA) and the European Medicines Agency (EMA) approved midostaurin (PKC412), a multiple kinase inhibitor that also inhibits KIT, for the treatment of advanced SM. Targeted therapy with midostaurin has shown efficacy in patients with advanced SM; however, overall survival in SM remains unsatisfactory, with a 3-year survival of 46% [11,12]. The new KIT inhibitor Avapritinib (BLU-285) also yielded very promising results in a recent study [13], but only a few patients (2 of 18) experienced a complete response. Allogeneic hematopoietic stem cell transplantation may represent a viable and potentially curative therapeutic option for advanced SM, although its definitive role as a treatment strategy remains to be determined [4]. Allogenic natural killer (NK) cells can efficiently eradicate myeloblasts but not malignant MCs in SM associated with acute myeloid leukemia (AML) [14]. Taken together, these findings underscore the need to better understand SM pathogenesis for the development of more efficient treatment strategies. New developments, including the use of next-generation sequencing (NGS) panels and the increasingly sensitive detection of the KIT D816V mutation have improved our understanding of SM pathogenesis. In this review, I discuss the important somatic molecular events that occur during the development of SM.
2. Molecular Events in the Development of SM
2.1. KIT
The interaction between KIT and its ligand, stem cell factor (SCF), plays an essential role in the regulation of MC maturation, proliferation, adhesion, chemotaxis, and survival [1,15,16]. Gain-of-function somatic mutations in the KIT tyrosine kinase domain, particularly the D816V mutation, have been identified in most adult SM patients, irrespective of the WHO SM subtype. Other, less common (<5%) somatic KIT mutations have also been identified in adult SM patients, including V560G, D815K, D816Y, D816F, D816H, and D820G. Interestingly, the KIT D816V mutation was detected in 84% of men and 75% of women analyzed (p < 0.001), which is consistent with generally worse outcomes observed for male patients with SM [17]. Generally, the KIT D816V mutation is less frequently detected in cases of childhood-onset mastocytosis than in adult patients (approximately 40% vs. >80%). Unlike classic SM, which is characterized by aberrant MC morphology, such as spindle shapes and hypogranulation, and associated with the KIT D816V mutation [1], a subgroup of patients (<10% of all SM) present with well-differentiated SM (WDSM), characterized by a mature MC morphology (round, fully granulated) and a reduced frequency of KIT D816V mutations compared with other forms of SM (29% vs. 93%) [18,19]. Chronic MCL is less aggressive than acute MCL, and chronic MCL is associated with morphologically mature MCs and is less frequently associated with KIT mutations than acute MCL [5]. Collectively, KIT mutations alone cannot explain the full clinical spectrum of SM. Moreover, the KIT D816V mutation burden does not correlate with clinical manifestations of ISM [20].
Although KIT mutations, particularly KIT D816V, have been considered to serve as key mutations in mastocytosis, an increasing body of data has indicated that other events (e.g., tropomyosin-related kinase [TRK]) may play important roles in the pathogenesis of mastocytosis (Table 1). New data suggested that KIT D816V is a late event in the pathogenesis of SM (Table 1) [8]. Moreover, animal studies have suggested that cooperating events are required for the effects of the KIT D816V mutation during the induction of SM [21,22]. Interestingly, recent data have cumulatively suggested that the effects of constitutive KIT signaling are dependent on the developmental stage of the cell, targeted by the gain-of-function mutation. In mastocytosis patients, mutations that targeting undifferentiated progenitors have been associated with result in multi-lineage involvement, and resulting in the expression of a severe systemic disease phenotype; in contrast, mutations that target committed MC progenitors or mature MCs alone lead to milder forms of the disease [10].
Determining whether mutated KIT is essential for the survival of malignant MCs remains an important goal. The identification of early cooperating events for KIT mutations may improve our understanding of the pathogenesis of SM, leading to more efficient treatments and improved outcomes for SM patients.
2.2. TRK (Tropomyosin-Related Kinase)
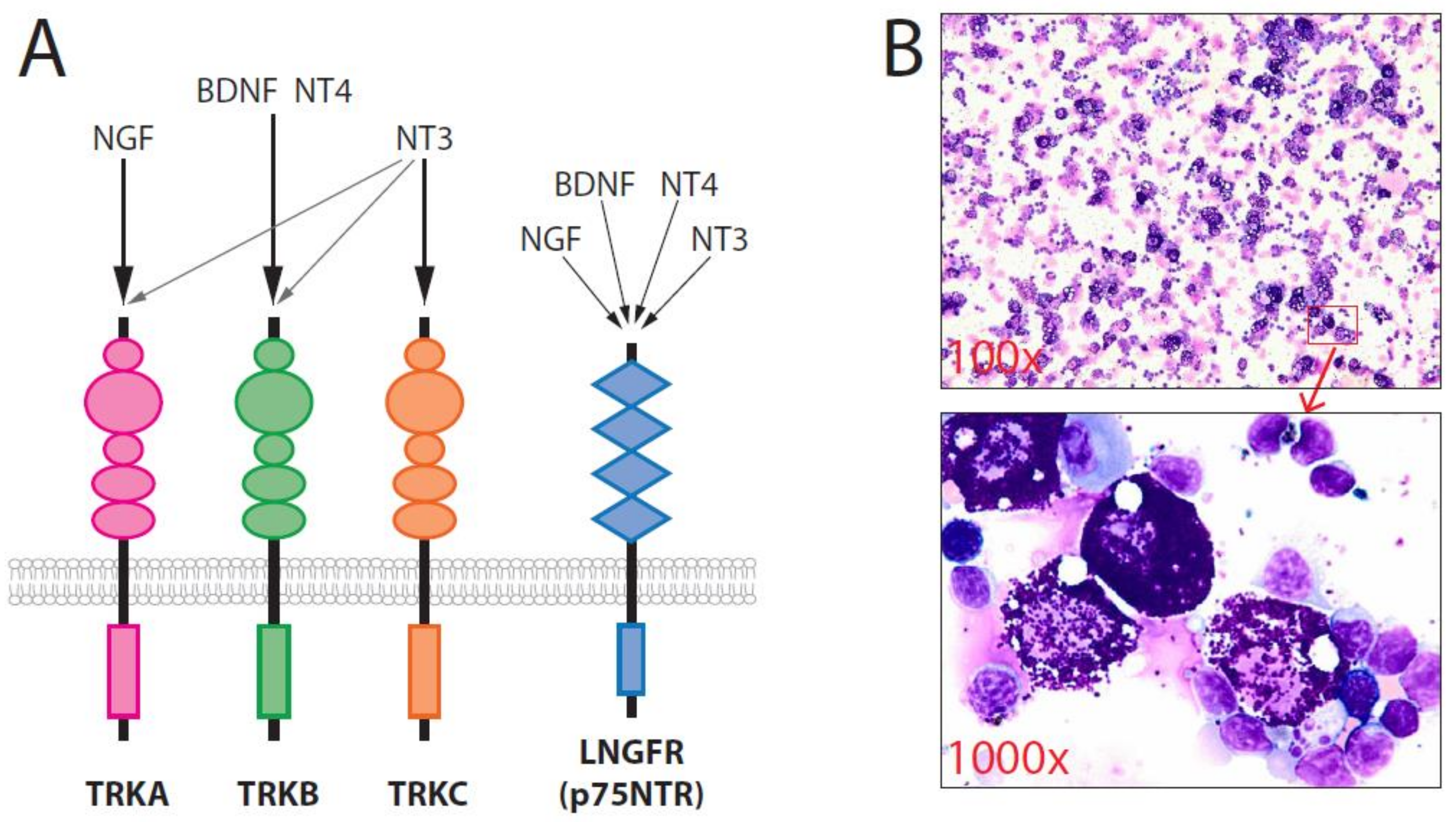
The neurotrophins (NTs), which include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), NT-3, and NT-4, play major roles in neuronal survival. NTs are unique because they utilize two different classes of receptors: the tropomyosin-related kinases (TRK), including TRKA, TRKB, and TRKC, and the low-affinity NGF receptor (LNGFR = p75NTR) [30,31]. NGF binds most specifically to TRKA, BDNF and NT-4 bind to TRKB, and NT-3 binds to TRKC. NT-3 can also induce the activation of TRKA and TRKB, in addition to TRKC. p75NTR can bind to all four NTs and regulate the affinity of TRK receptors for individual NTs (Figure 1). The promotion of neuronal cell survival by NT requires the activation of TRK, which triggers a Ras-dependent pathway that activates the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling pathways. TRK family members are expressed by several non-neural cell types and may also play crucial roles in the initiation, progression, and metastasis of many tumors in humans, including breast cancer. Aberrant TRKB signaling appears to be sufficient for the induction of tumorigenesis and metastasis [32], cell proliferation, and neoangiogenesis. In addition, some data have indicated the relevance of TRK receptors as prognostic factors [30]. Some TRK inhibitors are currently being tested in clinical trials, and entrectinib and Larotrectinib (LOXO-101) have been approved for use in patients with TRK fusion-positiv solid tumors, demonstrating that some human cancers are TRK dependent and that TRKs may be good targets for molecular therapy [33,34,35]. Notably, mutated TRK/TRK fusion is only found in 0.1–0.3% of human cancers [36].
The roles played by TRK receptors and their respective ligands during normal and malignant hematopoiesis are not yet well-understood. However, we and others have obtained evidence suggesting an important role for NT signaling in leukemogenesis and mastocytosis [30,38,39,40,41]. Moreover, TRK signaling has been suggested to be involved in other hematological malignancies, such as multiple myeloma. NTs have been known for many years to promote MC chemotaxis, maturation, and survival [40,42,43,44]. For instance, NGF can prevent apoptosis in cord-blood-derived human cultured MCs, acting synergistically with SCF [45]. NGF upregulates the expression of human MC factors, such as tryptase, and immature human MCs can be induced to assume a more mature phenotype in response to NGF treatment in vitro. NGF might have different biological effects at different stages of MC development [42]. Tryptase derived from human MCs can cleave pro-NGF, and mature NGF can be generated in response to tryptase activity [46], which may fundamentally modify the activities of pro-NGF/NGF [46]. Of note, MCs express the mRNA for all NTs and release active NGF, NT-3 (which can activate all TRKs), and NT-4 (which can activate TRKB) [47]. However, the role played by NT signaling in mastocytosis development remains largely unknown. An early report showed that NGF might be a major co-factor for the release of MC-mediators in SM without KIT D816V mutation [48]. Recently, Peng et al. demonstrated that patients with mastocytosis had enhanced NT levels and the elevated expression of TRKs on MCs in the skin and gut, and NGF increased the migration of KIT+ cells from the blood via TRKA, suggesting that TRK signaling might contribute to mastocytosis pathogenesis through autocrine and paracrine loops [40]. However, the precise contributions of TRKs to mastocytosis development and the underlying mechanisms remain largely unclear.
A clear phenotype-genotype correlation has been reported for TRKA and TRKB. Recently, we demonstrated for the first time that the activation of TRKB by BDNF and the activation of TRKA by NGF in murine hematopoietic stem cells (HSCs) and progenitor cells could efficiently induce a disease with striking similarities to human SM in vivo [29,37]. In 64 mice transplanted with retrovirally gene-modified primary HSC and progenitor cells, we observed SM development in the TRKA/NGF (3/7 = 43%) and TRKB/BDNF (12/17 = 71%) groups. SM primarily affected the spleen (Figure 1), liver, and bone marrow with multifocal, compact MC infiltrates. MCs mainly exhibited features of mature hypergranular MCs expressing transgene (e.g., TRKB/BDNF), c-Kit, tryptase, high-affinity receptors for IgE (FcεRI) and CD25. Most SM animals followed an indolent disease course. In >100 of our historical control mice transplanted in similar settings with different genes, including dTRKA, dLNGFR, FLT3 mutants, tCD34, and SV40 LT, no other mice developed SM [37]. These data strongly suggested that the activation of TRKA and TRKB by their ligands represents an important step in the promotion of mastocytosis. Our data indicated that both TRKA and TRKB activation were more potent than KIT D816V for the induction of SM [37], as the retroviral-mediated expression of KIT D816V failed to induce SM in transplanted mice [21]. In our models, the MC disease induced by TRKA/NGF and TRKB/BDNF was strikingly similar to human SM, particularly WDSM. The criteria for chronic human MCL were fulfilled in some mice. Although not all aspects of the human condition were reproduced in our models, the data indicated that TRKA and TRKB activation might play a critical role in the pathogenesis of SM without KIT D816V mutation, particularly in the development of WDSM or chronic MCL. Thus, targeting TRKs in such patients might represent a useful therapeutic strategy.
We found that the activation of TRKA or TRKB was involved in the development of resistance to KIT inhibitors mast cells with KIT mutations [29]. Targeting both TRK and KIT significantly prolonged the survival of mice xenotransplanted with HMC-1 cells compared with targeting KIT alone. The induction of TRKA activation in HMC-1 MCL cells that were resistant to KIT inhibition led to the reactivation of the MAPK/ERK signaling pathway and strong upregulation of early growth response 3 (EGR3, 1.269-fold), suggesting an important role for the MAPK-EGR3 axis in the development of resistance to KIT inhibition. Collectively, these data suggested that TRKA signaling may improve neoplastic MC fitness, which would explain, at least in part, why the results of treatment with KIT inhibitors alone in SM patients have been disappointing in most studies [1]. Because NGF is also expressed by bone marrow stromal cells [30], it might activate TRKA and protect MCs from cell death induced by KIT inhibition.
Taken together, we provided the first direct evidence to support SM development induced by the activation of TRKA or TRKB in HSC and progenitor cells in vivo [29,37]. Our data strongly supported the findings described by Peng et al. and their hypothesis [40], suggesting that TRKs have an important role in mastocytosis pathogenesis and the development of resistance to KIT inhibition.
We found that all TRK receptors were expressed on the surface of LAD2 cells, a commonly used MC line, and on primary malignant MCs derived from SM patients ([29] and own unpublished data). Because TRK expression can be identified in almost all patients with SM [40], together with the data from our group showing the induction of SM by TRKA and TRKB activation, TRK signaling may represent a disease-initiating molecular lesion. A synergistic anti-apoptotic effect has been observed for NGF/SCF and NT-3/SCF in human MCs [45,49]; thus, determining whether TRK signaling cooperates with KIT mutations in the induction of SM remains an important aspect for understanding mastocytosis development. One interesting question is whether the dysregulation of TRK signaling can modify the SM phenotype elicited by KIT mutations (i.e., promoting ISM vs. more aggressive SM subtypes). It is also important to investigate whether TRK is required for KIT signaling to induce mastocytosis.
2.3. mTOR Complexes
Mechanistic/mammalian target of rapamycin (mTOR) serves as a central regulator of cell growth and metabolism, forming two distinct complexes, mTORC1 and mTORC2, both of which phosphorylate multiple substrates [50,51]. The fine-tuning activity of mTOR complexes plays an important role in both the maintenance of HSCs and the suppression of leukemogenesis. The aberrant activation of the PI3K/mTOR pathway is a common feature of many cancers, including SM, and represents an attractive target for therapy [52]. Early studies have demonstrated that treatment with the mTOR inhibitor rapamycin depleted leukemic stem cells (LSCs) and restored the functions of normal HSCs [53]. Recent data showed that the depletion of mTORC1 activity failed to eliminate AML stem cells [54]. Interestingly, both Raptor and Rictor are important for B cell development, but the deletion of Raptor or Rictor does not alter progenitor cell survival or proliferation [54]. Recent data suggested that targeting Rictor/mTORC2 may be advantageous over the selective targeting of mTORC1 and may represent an attractive alternative approach for the eradication of tumor stem cells while sparing normal stem cell function [55,56,57]. Thus far, many inhibitors targeting the mTOR kinase have been developed; however, the therapeutic effects for SM have not been as robust as expected [50,56,58]. Moreover, very few reports have addressed the role played by the mTOR complexes in SM [52,59,60]. Although mTORC1 may contribute to MC survival, mTORC2 was only found to be critical for the homeostasis of neoplastic and dividing immature MCs [52]. However, no in vivo animal study has addressed the roles played by mTORC1 and mTORC2 in the pathogenesis of SM. Advances in our understanding of how mTOR signaling is involved in the homeostasis of normal HSCs and LSCs may lead to novel therapeutic approaches that may improve the clinical outcomes of patients with SM.
2.4. MAPK/ERK
ERK1 and ERK2 play key roles in cell survival, proliferation, cell adhesion, migration, and differentiation in many tissues [61]. ERK1/2 are required for the maintenance of HSCs and immature progenitor cells. The genetic disruption of ERK1/2 protected against the development of myeloproliferative neoplasms (MPN) in a mouse model [62]. ERK signaling appears to be important for the functions of MCs and might be involved in mastocytosis development [43,63]. Interestingly, dTRKA did not activate MAPK/ERK signaling, whereas the strong activation of ERK was observed following the TRKA activation by NGF. None of >20 C57BL/6J mice transplanted with dTRKA-modified primary Lin-cells developed SM, in contrast with the development of SM in 43% of mice transplanted with TRKA/NGF, suggesting that ERK may be important for mastocytosis development. Because the role of ERK1 and ERK2 in MCs and SM development has not been well investigated, the investigation of the roles played by ERK1 and ERK2 in SM development mediated by TRK and KIT signaling should be explored to determine whether ERK1, ERK2, or both isoforms are required for SM development.
2.5. Other Alterations/Mutations
More recently, new mutations have been identified in mastocytoses, such as TET2, ASLX1, SRSF2, JAK2 V617F, Nras, and ETNK1 (ethanolamine kinase 1) [3,16,64,65]. Interestingly, one study suggested that the overall survival was adversely affected by mutations in SRSF2, ASXL1, and RUNX1 [64]. In addition, some signaling pathways have been suggested to be important for the pathogenesis of SM, such as signal transducer and activator of transcription 5 (STAT5) and AKT [43,63]. However, these newly identified mutations and signaling pathways are not specific to mastocytosis [66], and the pathogenetic roles, prognostic, and therapeutic impact of the majority of these mutations and pathways remain to be determined [3]. A recently published study demonstrated that co-expression of kitD814V mutation (homolog to human KIT D816V) and loss of TET2 led to a more aggressive phenotype in the skin and the digestive tract than KIT D816 V alone [67]. However, increased mast cell accumulation in other organ systems was not observed, and the co-existence of both mutations was insufficient to confer IL-3 independence to BMMCs (mast cells from bone marrow progenitors), suggesting that additional alterations are still required. Loss-of-heterozygosity of SETD2 was detected in 27/57 (47%) SM patients [68], suggesting a role of SETD2 deficiency in the pathogenesis of SM. Moreover, mutations in the TP53 gene are not common in SM but might cooperate with KIT D816V to initiate SM [69]. Identifying these genetic and /epigenetic alterations and understanding their interactions and the molecular mechanisms involved in mastocytosis is of the utmost importance for developing rationally targeted therapies.
2.6. Cytogenetic Abnormalities
So far, there are only a few reports of cytogenetic analyses in SM patients. Interestingly, cytogenetic abnormalities are highly restricted to patients with advanced SM in two recent publications [17,70], suggesting that they are unlikely to be primary events in the pathogenesis of SM but rather that these abnormalities promote progression to advanced SM.
3. Cell Origins of SM
It is well known that MCs are derived from HSCs. The experiments from Gall`s group strongly suggest that adult MC progenitors are derived directly from multipotential progenitors [71]. In contrast to transplantation with primary HSCs and progenitor cells, C3H/HeJ mice transplanted with TRKB- and BDNF-modified 32D cells (murine myeloid progenitors) only developed AML, with no signs of increased numbers of mastocyte [37]. The finding that mastocytosis is induced only when TRKB is activated in HSCs and progenitor cells strongly support the accepted view that MCs are derived from HSCs. There are increasing reports of the co-occurrence of SM with hematological malignancies. Nearly all patients with KIT D816V-mutated myelodysplastic syndromes (MDS), MPN, or overlapping MDS/MPN exhibited concurrent SM, whereas 44% of patients with AML and KIT D816V showed evidence of SM at some point in their disease course [72]. This suggests that a common cell origin for SM and other hematological malignancies might exist. In a single-cell analysis study, Grootens et al. [73] demonstrated that the KIT D816V mutation in SM can be found in cells throughout the hematopoietic landscape, supporting the view a malignant clone may arise in the HSC compartment. Whether other hematological progenitors can be transformed and initiate SM remains to be determined.
4. Impacts on the Treatment of SM
More efficient treatment strategies are required to improve the outcomes of patients with SM [74]. Avapritinib is a more potent KIT inhibitor than midostaurin and appears to induce complete responses in more patients [13]. Developing more potent KIT inhibitors for the treatment of SM remains an important goal. The potent new inhibitor ripretinib (DCC-2618) broadly inhibits primary and drug-resistant KIT/PDGFRA mutants [75] and is currently being tested in clinical trials. In 2020 ripretinib was first approved in the USA for the treatment of adults with advanced gastrointestinal stromal tumors (GIST) who had already undergone prior treatments with ≥3 kinase inhibitors. Because the KIT D816V mutation alone cannot induce SM in a transplantation model, however, it is obvious that targeting KIT alone will not control the disease in the long term. Therefore, there is an urgent need to develop novel combined therapies for use in SM patients. A clinical trial examining the combination of KIT inhibition and chemotherapy could provide insightful new data. Other combined therapies including KIT and TRK inhibition [29], KIT and proteasome inhibition [68], and those including donor lymphocyte infusion [76] might be efficient SM treatment strategies.
In addition, it remains to be determined whether chimeric antigen receptor (CAR)-T cell therapies are also effective for the treatment of SM. CARs are unique receptors that are generated to target a specific tumor antigen to functionally reprogram T lymphocytes. CAR-T cell therapy has yielded impressive effects in patients with B-lineage acute lymphoblastic leukemia [77], but to date, there have been no reports of the development of CAR-T cell therapy for SM. Because CD123 is aberrantly expressed on neoplastic MCs [78], and CD123 CAR-T cells are now being intensively tested for treatment of AML in several trials [79], testing CD123 CAR-T cells might be a first step in the development of CAR-T cell therapy for SM.
5. Conclusions
SM is a rare hematological malignancy with complex diagnostic and clinical classifications [5,80]. SM research is lagging behind research investigating other common hematological malignancies such as AML and chronic myeloid leukemia, and fewer scientists and physicians are working in the field of SM; thus it is important to strengthen our collaborations to improve understanding of its pathogenesis. A better understanding of the mechanisms underlying the development of SM may yield improved molecular therapies for its treatment. Patient-specific KIT D816V induced pluripotent stem cells (so-called iPSCs) from patients with ASM and MCL represent a patient-specific SM disease model for mechanistic and drug discovery studies. The angiokinase inhibitor nintedanib which targets VEGFR, PDGFR, and FGFR is approved for the treatment of non-small-cell lung cancer, idiopathic pulmonary fibrosis, and other lung diseases by the FDA and EMA was identified as a novel KIT D816V inhibitor using this model [81]. These results suggest nintedanib as a new drug candidate for targeted therapy in patients with advanced SM. Different prognostic scoring systems and genetic biomarkers have been reported [82,83,84,85,86]. These can be used to predict survival outcomes and guide therapy decisions. With novel targeted therapies entering clinical trials, there is now hope for SM patients, particularly those with advanced SM who frequently have impaired quality of life or a poor prognosis. Interestingly, bone marrow MCs lack angiotensin-converting enzyme 2 receptors, and concurrent coronavirus disease 2019 (COVID-19) in SM patients reportedly does not impact MC activation symptoms [87]. Moreover, patients with CM and SM with MC activation symptoms and anaphylaxis reportedly tolerate mRNA COVID-19 vaccination well [88]. We hope that the quality of life of SM patients will be further improved [89].
Author Contributions
Z.L. contributed to all steps of creating this review. Author has read and agreed to the published version of the manuscript.
Funding
The research conducted in the laboratory of ZL was supported by Deutsche Forschungsgemeinschaft (grants Li 1608/2-1 and Li 1608/5-1), the Deutsche José Carreras Leukämie-Stiftung (grants 13/22 and 21 R/2017), the Deutsche Krebshilfe (grant 108245), and Alfred & Angelika Gutermuth-Stiftung (projects 2016/3, 2017/3, 2018/3, 2019/2, and 2020/4).
Acknowledgments
The author is grateful to Min Yang (Hannover Medical School) for preparing Figure 1.
Conflicts of Interest
The author declares no conflict of interest.
References
- Theoharides, T.C.; Valent, P.; Akin, C. Mast Cells, Mastocytosis, and Related Disorders. N. Engl. J. Med. 2015, 373, 1885–1886. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Advances in the Classification and Treatment of Mastocytosis: Current Status and Outlook toward the Future. Cancer Res. 2017, 77, 1261–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2015, 90, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Arock, M.; Kluin-Nelemans, H.C.; Reiter, A.; Sperr, W.R.; George, T.; Horny, H.P.; Hartmann, K.; Sotlar, K.; Damaj, G.; et al. Advanced systemic mastocytosis: From molecular and genetic progress to clinical practice. Haematologica 2016, 101, 1133–1143. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Pardanani, A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am. J. Hematol. 2021, 96, 508–525. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Muller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Giles, F.J.; le Coutre, P.D.; Muller, M.C.; Reiter, A.; Santanastasio, H.; Leung, M.; Novick, S.; Kantarjian, H.M. Nilotinib in patients with systemic mastocytosis: Analysis of the phase 2, open-label, single-arm nilotinib registration study. J. Cancer Res. Clin. Oncol. 2015, 141, 2047–2060. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am. J. Hematol. 2016, 91, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; Akin, C.; Hartmann, K.; Valent, P.; Reiter, A. Practical management of adverse events in patients with advanced systemic mastocytosis receiving midostaurin. Expert Opin. Biol. 2021, 1–12. [Google Scholar] [CrossRef]
- Rapid Responses to Avapritinib (BLU-285) in Mastocytosis. Cancer Discov. 2018, 8, 133.
- Ustun, C.; Williams, S.; Skendzel, S.; Kodal, B.; Arock, M.; Gotlib, J.; Vallera, D.A.; Cooley, S.; Felices, M.; Weisdorf, D.; et al. Allogeneic NK cells eradicate myeloblasts but not neoplastic mast cells in systemic mastocytosis associated with acute myeloid leukemia. Am. J. Hematol. 2017, 92, E66–E68. [Google Scholar] [CrossRef] [Green Version]
- Hoermann, G.; Gleixner, K.V.; Dinu, G.E.; Kundi, M.; Greiner, G.; Wimazal, F.; Hadzijusufovic, E.; Mitterbauer, G.; Mannhalter, C.; Valent, P.; et al. The KIT D816V allele burden predicts survival in patients with mastocytosis and correlates with the WHO type of the disease. Allergy 2014, 69, 810–813. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Mast cells as a unique hematopoietic lineage and cell system: From Paul Ehrlich’s visions to precision medicine concepts. Theranostics 2020, 10, 10743–10768. [Google Scholar] [CrossRef]
- Kluin-Nelemans, H.C.; Jawhar, M.; Reiter, A.; van Anrooij, B.; Gotlib, J.; Hartmann, K.; Illerhaus, A.; Oude Elberink, H.N.G.; Gorska, A.; Niedoszytko, M.; et al. Cytogenetic and molecular aberrations and worse outcome for male patients in systemic mastocytosis. Theranostics 2021, 11, 292–303. [Google Scholar] [CrossRef]
- Akin, C.; Fumo, G.; Yavuz, A.S.; Lipsky, P.E.; Neckers, L.; Metcalfe, D.D. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood 2004, 103, 3222–3225. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Teodosio, C.; Sanchez, M.L.; Nunez, R.; Prados, A.; Aldanondo, I.; Sanchez, L.; Dominguez, M.; Botana, L.M.; et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: A prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef]
- Broesby-Olsen, S.; Kristensen, T.; Vestergaard, H.; Brixen, K.; Moller, M.B.; Bindslev-Jensen, C. Mastocytosis Centre Odense University, H. KIT D816V mutation burden does not correlate to clinical manifestations of indolent systemic mastocytosis. J. Allergy Clin. Immunol. 2013, 132, 723–728. [Google Scholar] [CrossRef]
- Xiang, Z.; Kreisel, F.; Cain, J.; Colson, A.; Tomasson, M.H. Neoplasia driven by mutant c-KIT is mediated by intracellular, not plasma membrane, receptor signaling. Mol. Cell. Biol. 2007, 27, 267–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappulla, J.P.; Dubreuil, P.; Desbois, S.; Letard, S.; Hamouda, N.B.; Daeron, M.; Delsol, G.; Arock, M.; Liblau, R.S. Mastocytosis in mice expressing human Kit receptor with the activating Asp816Val mutation. J. Exp. Med. 2005, 202, 1635–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, M.; Nedoszytko, B.; Gorska, A.; Zawrocki, A.; Sobjanek, M.; Kozlowski, D. Mastocytosis in children and adults: Clinical disease heterogeneity. Arch. Med. Sci. 2012, 8, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Wedeh, G.; Herrmann, H.; Bibi, S.; Cerny-Reiterer, S.; Sadovnik, I.; Blatt, K.; Hadzijusufovic, E.; Jeanningros, S.; Blanc, C.; et al. A new human mast cell line expressing a functional IgE receptor converts to tumorigenic growth by KIT D816V transfection. Blood 2014, 124, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Mayerhofer, M.; Gleixner, K.V.; Hoelbl, A.; Florian, S.; Hoermann, G.; Aichberger, K.J.; Bilban, M.; Esterbauer, H.; Krauth, M.T.; Sperr, W.R.; et al. Unique effects of KIT D816V in BaF3 cells: Induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J. Immunol. 2008, 180, 5466–5476. [Google Scholar] [CrossRef] [Green Version]
- Balci, T.B.; Prykhozhij, S.V.; Teh, E.M.; Da’as, S.I.; McBride, E.; Liwski, R.; Chute, I.C.; Leger, D.; Lewis, S.M.; Berman, J.N. A transgenic zebrafish model expressing KIT-D816V recapitulates features of aggressive systemic mastocytosis. Br. J. Haematol. 2014, 167, 48–61. [Google Scholar] [CrossRef]
- Gerbaulet, A.; Wickenhauser, C.; Scholten, J.; Peschke, K.; Drube, S.; Horny, H.P.; Kamradt, T.; Naumann, R.; Muller, W.; Krieg, T.; et al. Mast cell hyperplasia, B-cell malignancy, and intestinal inflammation in mice with conditional expression of a constitutively active kit. Blood 2011, 117, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Kitayama, H.; Tsujimura, T.; Matsumura, I.; Oritani, K.; Ikeda, H.; Ishikawa, J.; Okabe, M.; Suzuki, M.; Yamamura, K.; Matsuzawa, Y.; et al. Neoplastic transformation of normal hematopoietic cells by constitutively activating mutations of c-kit receptor tyrosine kinase. Blood 1996, 88, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Pan, Z.; Huang, K.; Busche, G.; Feuerhake, F.; Chaturvedi, A.; Nie, D.; Heuser, M.; Thol, F.; von Neuhoff, N.; et al. Activation of TRKA receptor elicits mastocytosis in mice and is involved in the development of resistance to KIT-targeted therapy. Oncotarget 2017, 8, 73871–73883. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Beutel, G.; Rhein, M.; Meyer, J.; Koenecke, C.; Neumann, T.; Yang, M.; Krauter, J.; von Neuhoff, N.; Heuser, M.; et al. High-affinity neurotrophin receptors and ligands promote leukemogenesis. Blood 2009, 113, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Mitre, M.; Mariga, A.; Chao, M.V. Neurotrophin signalling: Novel insights into mechanisms and pathophysiology. Clin. Sci. 2017, 131, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Douma, S.; Van Laar, T.; Zevenhoven, J.; Meuwissen, R.; Van Garderen, E.; Peeper, D.S. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 2004, 430, 1034–1039. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Goldman, D.A.; Hechtman, J.F.; Benayed, R.; Schram, A.M.; Cocco, E.; Shifman, S.; Gong, Y.; Kundra, R.; Solomon, J.P.; et al. TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin. Cancer Res. 2020, 26, 1624–1632. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Huang, K.; Busche, G.; Ganser, A.; Li, Z. Activation of TRKB receptor in murine hematopoietic stem/progenitor cells induced mastocytosis. Blood 2014, 124, 1196–1197. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dullmann, J.; Schiedlmeier, B.; Schmidt, M.; von Kalle, C.; Meyer, J.; Forster, M.; Stocking, C.; Wahlers, A.; Frank, O.; et al. Murine leukemia induced by retroviral gene marking. Science 2002, 296, 497. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.; Rhein, M.; Schiedlmeier, B.; Kustikova, O.; Rudolph, C.; Kamino, K.; Neumann, T.; Yang, M.; Wahlers, A.; Fehse, B.; et al. Remarkable leukemogenic potency and quality of a constitutively active neurotrophin receptor, deltaTrkA. Leukemia 2007, 21, 2171–2180. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.M.; Maintz, L.; Allam, J.P.; Raap, U.; Gutgemann, I.; Kirfel, J.; Wardelmann, E.; Perner, S.; Zhao, W.; Fimmers, R.; et al. Increased circulating levels of neurotrophins and elevated expression of their high-affinity receptors on skin and gut mast cells in mastocytosis. Blood 2013, 122, 1779–1788. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Schwaller, J.; Kutok, J.; Cain, D.; Aster, J.C.; Williams, I.R.; Gilliland, D.G. Signal transduction and transforming properties of the TEL-TRKC fusions associated with t(12;15)(p13;q25) in congenital fibrosarcoma and acute myelogenous leukemia. Embo. J. 2000, 19, 1827–1838. [Google Scholar] [CrossRef] [Green Version]
- Welker, P.; Grabbe, J.; Grutzkau, A.; Henz, B.M. Effects of nerve growth factor (NGF) and other fibroblast-derived growth factors on immature human mast cells (HMC-1). Immunology 1998, 94, 310–317. [Google Scholar]
- Sawada, J.; Itakura, A.; Tanaka, A.; Furusaka, T.; Matsuda, H. Nerve growth factor functions as a chemoattractant for mast cells through both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling pathways. Blood 2000, 95, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Okayama, Y.; Kawakami, T. Development, migration, and survival of mast cells. Immunol Res. 2006, 34, 97–115. [Google Scholar] [CrossRef]
- Kanbe, N.; Kurosawa, M.; Miyachi, Y.; Kanbe, M.; Saitoh, H.; Matsuda, H. Nerve growth factor prevents apoptosis of cord blood-derived human cultured mast cells synergistically with stem cell factor. Clin. Exp. Allergy 2000, 30, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Spinnler, K.; Frohlich, T.; Arnold, G.J.; Kunz, L.; Mayerhofer, A. Human tryptase cleaves pro-nerve growth factor (pro-NGF): Hints of local, mast cell-dependent regulation of NGF/pro-NGF action. J. Biol. Chem. 2011, 286, 31707–31713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaper, S.D. Nerve growth factor: A neuroimmune crosstalk mediator for all seasons. Immunology 2017, 151. [Google Scholar] [CrossRef]
- Kurosawa, M.; Inamura, H.; Amano, H.; Kanbe, N.; Nagata, H.; Nagai, H.; Furukawa, S.; Miyachi, Y. Nerve growth factor release with mast-cell-derived mediators in a patient with systemic mastocytosis after middle-wave ultraviolet irradiation. Allergy 1999, 54, 994–998. [Google Scholar] [CrossRef]
- Lorentz, A.; Hoppe, J.; Worthmann, H.; Gebhardt, T.; Hesse, U.; Bienenstock, J.; Bischoff, S.C. Neurotrophin-3, but not nerve growth factor, promotes survival of human intestinal mast cells. Neurogastroenterol. Motil. 2007, 19, 301–308. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Smrz, D.; Kim, M.S.; Zhang, S.; Mock, B.A.; Smrzova, S.; DuBois, W.; Simakova, O.; Maric, I.; Wilson, T.M.; Metcalfe, D.D.; et al. mTORC1 and mTORC2 differentially regulate homeostasis of neoplastic and non-neoplastic human mast cells. Blood 2011, 118, 6803–6813. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T.; Sugiyama, N.; Soga, T.; Araki, K.; Yamamura, K.; Hirao, A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Invest. 2012, 122, 2114–2129. [Google Scholar] [CrossRef]
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; Guan, K.L.; Morrison, S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Yang, Y.; Hua, C.; Xu, S.; Zhou, M.; Guo, H.; Wang, N.; Zhao, X.; Huang, L.; Yu, F.; et al. Rictor has a pivotal role in maintaining quiescence as well as stemness of leukemia stem cells in MLL-driven leukemia. Leukemia 2017, 31, 414–422. [Google Scholar] [CrossRef]
- Lee, K.; Nam, K.T.; Cho, S.H.; Gudapati, P.; Hwang, Y.; Park, D.S.; Potter, R.; Chen, J.; Volanakis, E.; Boothby, M. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J. Exp. Med. 2012, 209, 713–728. [Google Scholar] [CrossRef]
- Parikh, S.A.; Kantarjian, H.M.; Richie, M.A.; Cortes, J.E.; Verstovsek, S. Experience with everolimus (RAD001), an oral mammalian target of rapamycin inhibitor, in patients with systemic mastocytosis. Leuk. Lymphoma 2010, 51, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Gabillot-Carre, M.; Lepelletier, Y.; Humbert, M.; de Sepuvelda, P.; Hamouda, N.B.; Zappulla, J.P.; Liblau, R.; Ribadeau-Dumas, A.; Machavoine, F.; Letard, S.; et al. Rapamycin inhibits growth and survival of D816V-mutated c-kit mast cells. Blood 2006, 108, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Kuehn, H.S.; Metcalfe, D.D.; Gilfillan, A.M. Activation and function of the mTORC1 pathway in mast cells. J. Immunol. 2008, 180, 4586–4595. [Google Scholar] [CrossRef]
- Chan, G.; Gu, S.; Neel, B.G. Erk1 and Erk2 are required for maintenance of hematopoietic stem cells and adult hematopoiesis. Blood 2013, 121, 3594–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staser, K.; Park, S.J.; Rhodes, S.D.; Zeng, Y.; He, Y.Z.; Shew, M.A.; Gehlhausen, J.R.; Cerabona, D.; Menon, K.; Chen, S.; et al. Normal hematopoiesis and neurofibromin-deficient myeloproliferative disease require Erk. J. Clin. Invest. 2013, 123, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Niedoszytko, M.; Oude Elberink, J.N.; Bruinenberg, M.; Nedoszytko, B.; de Monchy, J.G.; te Meerman, G.J.; Weersma, R.K.; Mulder, A.B.; Jassem, E.; van Doormaal, J.J. Gene expression profile, pathways, and transcriptional system regulation in indolent systemic mastocytosis. Allergy 2011, 66, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Zblewski, D.; Patnaik, M.; Ketterling, R.P.; Chen, D.; Hanson, C.A.; Tefferi, A.; Pardanani, A. Novel recurrent mutations in ethanolamine kinase 1 (ETNK1) gene in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia. Blood Cancer J. 2015, 5, e275. [Google Scholar] [CrossRef] [Green Version]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef] [Green Version]
- De Vita, S.; Schneider, R.K.; Garcia, M.; Wood, J.; Gavillet, M.; Ebert, B.L.; Gerbaulet, A.; Roers, A.; Levine, R.L.; Mullally, A.; et al. Loss of function of TET2 cooperates with constitutively active KIT in murine and human models of mastocytosis. PLoS ONE 2014, 9, e96209. [Google Scholar] [CrossRef]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2018, 32, 139–148. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Miura, H.; Inagaki, H.; Shinkai, Y.; Kato, A.; Kato, T.; Hamada-Tsutsumi, S.; Tanaka, M.; Kudo, K.; Yoshikawa, T.; et al. An aggressive systemic mastocytosis preceded by ovarian dysgerminoma. BMC Cancer 2020, 20, 1162. [Google Scholar] [CrossRef]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lubke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.P.; Sotlar, K.; et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer 2018, 57, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Grimbaldeston, M.A.; Tsai, M.; Weissman, I.L.; Galli, S.J. Identification of mast cell progenitors in adult mice. Proc. Natl. Acad. Sci. USA 2005, 102, 11408–11413. [Google Scholar] [CrossRef] [Green Version]
- Craig, J.W.; Hasserjian, R.P.; Kim, A.S.; Aster, J.C.; Pinkus, G.S.; Hornick, J.L.; Steensma, D.P.; Coleman Lindsley, R.; DeAngelo, D.J.; Morgan, E.A. Detection of the KIT(D816V) mutation in myelodysplastic and/or myeloproliferative neoplasms and acute myeloid leukemia with myelodysplasia-related changes predicts concurrent systemic mastocytosis. Mod. Pathol. 2020, 33, 1135–1145. [Google Scholar] [CrossRef]
- Grootens, J.; Ungerstedt, J.S.; Ekoff, M.; Ronnberg, E.; Klimkowska, M.; Amini, R.M.; Arock, M.; Soderlund, S.; Mattsson, M.; Nilsson, G.; et al. Single-cell analysis reveals the KIT D816V mutation in haematopoietic stem and progenitor cells in systemic mastocytosis. EBioMedicine 2019, 43, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Reiter, A.; George, T.I.; Gotlib, J. New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood 2020, 135, 1365–1376. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751. [Google Scholar] [CrossRef]
- Hagglund, H.; Yavuz, A.S.; Dreimane, A.; Malm, C.; Sundin, A.; Sander, B.; Nilsson, G. Graft-versus-mastocytosis effect after donor lymphocyte infusion: Proof of principle. Eur. J. Haematol. 2021, 106, 290–293. [Google Scholar] [CrossRef]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.; Chen, D.; Kimlinger, T.K.; Finke, C.; Zblewski, D.; Patnaik, M.M.; Reichard, K.K.; Rowinsky, E.; Hanson, C.A.; et al. Aberrant expression of CD123 (interleukin-3 receptor-alpha) on neoplastic mast cells. Leukemia 2015, 29, 1605–1608. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef]
- Radia, D.H.; Green, A.; Oni, C.; Moonim, M. The clinical and pathological panoply of systemic mastocytosis. Br. J. Haematol. 2020, 188, 623–640. [Google Scholar] [CrossRef]
- Toledo, M.A.S.; Gatz, M.; Sontag, S.; Gleixner, K.V.; Eisenwort, G.; Feldberg, K.; Hamouda, A.E.I.; Kluge, F.; Guareschi, R.; Rossetti, G.; et al. Nintedanib Targets KIT D816V Neoplastic Cells Derived from Induced Pluripotent Stem cells of Systemic Mastocytosis. Blood 2020, 137, 2070–2084. [Google Scholar] [CrossRef] [PubMed]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Oude Elberink, J.N.G.; Gorska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International prognostic scoring system for mastocytosis (IPSM): A retrospective cohort study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Alvarez-Twose, I.; Shoumariyeh, K.; Naumann, N.; Lubke, J.; Perkins, C.; Munoz-Gonzalez, J.I.; Meggendorfer, M.; Kennedy, V.; et al. MARS: Mutation-Adjusted Risk Score for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2019, 37, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gonzalez, J.I.; Alvarez-Twose, I.; Jara-Acevedo, M.; Zanotti, R.; Perkins, C.; Jawhar, M.; Sperr, W.R.; Shoumariyeh, K.; Schwaab, J.; Greiner, G.; et al. Proposed global prognostic score for systemic mastocytosis: A retrospective prognostic modelling study. Lancet Haematol. 2021, 8, e194–e204. [Google Scholar] [CrossRef]
- Greiner, G.; Sprinzl, B.; Gorska, A.; Ratzinger, F.; Gurbisz, M.; Witzeneder, N.; Schmetterer, K.G.; Gisslinger, B.; Uyanik, G.; Hadzijusufovic, E.; et al. Hereditary alpha tryptasemia is a valid genetic biomarker for severe mediator-related symptoms in mastocytosis. Blood 2021, 137, 238–247. [Google Scholar] [CrossRef]
- Munoz-Gonzalez, J.I.; Alvarez-Twose, I.; Jara-Acevedo, M.; Henriques, A.; Vinas, E.; Prieto, C.; Sanchez-Munoz, L.; Caldas, C.; Mayado, A.; Matito, A.; et al. Frequency and prognostic impact of KIT and other genetic variants in indolent systemic mastocytosis. Blood 2019, 134, 456–468. [Google Scholar] [CrossRef]
- Giannetti, M.P.; Weller, E.; Alvarez-Twose, I.; Torrado, I.; Bonadonna, P.; Zanotti, R.; Dwyer, D.F.; Foer, D.; Akin, C.; Hartmann, K.; et al. COVID-19 infection in patients with mast cell disorders including mastocytosis does not impact mast cell activation symptoms. J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Rama, T.A.; Moreira, A.; Castells, M. mRNA COVID-19 vaccine is well tolerated in patients with cutaneous and systemic mastocytosis with mast cell activation symptoms and anaphylaxis. J. Allergy Clin. Immunol. 2021, 147, 877–878. [Google Scholar] [CrossRef]
- Hartmann, K.; Gotlib, J.; Akin, C.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Menssen, H.D.; Redhu, S.; Knoll, S.; et al. Midostaurin improves quality of life and mediator-related symptoms in advanced systemic mastocytosis. J. Allergy Clin. Immunol. 2020, 146, 356–366. [Google Scholar] [CrossRef]
Figure 1.
(A) The neurotrophin family has four members: NGF, BDNF, NT-3, and NT-4. BDNF, NT-3, and NT-4 can activate TRKB. (B) Overexpression of wild-type TRKB/BDNF in murine primary hematopoietic stem/progenitor cells induced SM. Cytospins showing strong infiltration of mature MCs in the spleen [37].
Figure 1.
(A) The neurotrophin family has four members: NGF, BDNF, NT-3, and NT-4. BDNF, NT-3, and NT-4 can activate TRKB. (B) Overexpression of wild-type TRKB/BDNF in murine primary hematopoietic stem/progenitor cells induced SM. Cytospins showing strong infiltration of mature MCs in the spleen [37].

{kind=link}
Table 1.
Data supporting the important roles of events other than KIT mutations in mastocytosis pathogenesis.
Table 1.
Data supporting the important roles of events other than KIT mutations in mastocytosis pathogenesis.
| Data and References | |
|---|---|
| 1 | Although both childhood- and adult-onset mastocytosis are associated with activating KIT mutations, the clinical presentation and outcomes of these two conditions differ (a skin-limited disease that spontaneously regresses with age vs. persistent, multi-organ involvement, often with a concurrent non-MC hematologic neoplasm) [3,23]. KIT mutations alone cannot explain the full clinical spectrum of SM. |
| 2 | KIT D816V does not activate mast cells to release proinflammatory mediators [24]. |
| 3 | KIT D816V mutation appears to be a late event in the pathogenesis of mastocytosis [8]. |
| 4 | KIT D816V is a weak oncogene. For example, BaF3 cells with conditional expression of KIT D816V did not form tumors in nude mice [25]. |
| 5 | KIT D816V is thought to promote MC differentiation and maturation rather than MC proliferation [5]. |
| 6 | The retroviral-mediated expression of KIT D816V failed to induce SM in transplanted animals [21]. Only 29% of transgenic mice expressing human KIT D816V developed mastocytosis at an old age (>12 months) [22]. In addition, 50% of transgenic zebrafish expressing KIT D616V demonstrated a myeloproliferative disease phenotype, including features of ASM [26]. Although mastocytosis was observed in all transgenic mice expressing murine KitD814V (homolog of the human D816V mutant) in another model, the disease occurred significantly later and progressed slower when KitD814V was expressed in mature MCs [27]. Mastocytosis was not observed at all in transgenic mice expressing KitD814V in an earlier study [28]. |
| 7 | Targeting KIT in SM may produce a substantial reduction in MC burden in some patients, however, treatment with KIT inhibitors alone, thus far, has been disappointing in most patients with mastocytosis [1], likely due to the development of resistance to kinase inhibitors [4,29]. A recently published study demonstrated a therapeutic benefit in patients with advanced SM associated with the use of midostaurin (PKC412), a multiple kinase inhibitor that inhibits KIT [11]; however, overall survival at 3 years was only 46%. Few, if any, patients achieved complete remission. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Z. New Insights into the Pathogenesis of Systemic Mastocytosis. Int. J. Mol. Sci. 2021, 22, 4900. https://doi.org/10.3390/ijms22094900
AMA Style
Li Z. New Insights into the Pathogenesis of Systemic Mastocytosis. International Journal of Molecular Sciences. 2021; 22(9):4900. https://doi.org/10.3390/ijms22094900
Chicago/Turabian StyleLi, Zhixiong. 2021. "New Insights into the Pathogenesis of Systemic Mastocytosis" International Journal of Molecular Sciences 22, no. 9: 4900. https://doi.org/10.3390/ijms22094900
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.