1. Introduction
In eukaryotic cells, secretion is a fundamental process for delivery of newly synthesized materials to the plasma membrane (PM) or extracellular space to achieve various biological functions. The plant secretory trafficking network is of vital importance in many growth and development processes, including cell morphogenesis, cell wall biosynthesis, hormone signaling, cell polarity formation, as well as response to biotic and abiotic stress [
1,
2,
3,
4]. Proteins or lipids are packaged in membrane-bound vesicles that are targeted to specific PM domains for exocytosis. Vesicle trafficking and delivery are highly-choreographed multistep processes requiring a diverse set of proteins and protein–protein interactions to ensure precise spatiotemporal regulation of cargo secretion [
5,
6]. Typically, secretory vesicles are transported to their destination membrane along cytoskeletal tracks and powered by motor proteins. In some cell types, vesicles are trapped or captured by a cortical cytoskeletal meshwork. Once at the PM, secretory vesicle tethering, docking, and membrane fusion occur in sequential fashion. During tethering, proteins from both the vesicle surface and the PM form a complex to stabilize vesicles near the secretion site; another set of proteins including soluble
N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) and SNARE-related proteins further bring vesicles in close proximity during the docking stage; finally, the lipid bilayers of vesicle membrane and PM fuse to allow cargo release and/or incorporation of membrane-associated proteins [
7,
8,
9]. Although the exocytic process and molecular machinery are well characterized in yeast and animal models, emerging studies in plants reveal similar exocytic trafficking systems and a range of evolutionarily-conserved and plant-specific molecular regulators [
10,
11,
12,
13].
Plant cells are encased within a carbohydrate-rich cell wall comprised mainly of cellulose microfibrils as the major load-bearing component. Cellulose is a β-1,4-linked glucan chain that is synthesized by cellulose synthase (CESA) enzymes located at the PM and multiple CESAs are arrayed in large, multimeric rosette-shaped cellulose synthase complexes (CSCs) [
14]. CSCs are thought to be assembled in the Golgi and delivered to the PM by post-Golgi vesicles [
15,
16]. Some of the CSC-containing vesicles are referred to as small CESA compartments (SmaCCs) or microtubule-associated CESA compartments (MASCs) [
17,
18]. CSCs are preferentially delivered to PM sites that coincide with cortical microtubules [
18]. Furthermore, CSCs at the PM are linked to underlying cortical microtubules via microtubule-associated proteins such as CELLULOSE SYNTHASE INTERACTIVE 1/POM2 (CSI1/POM2) and move in linear trajectories at a constant speed to produce cellulose microfibrils [
19,
20,
21,
22]. Although cortical microtubules define their trajectories, the motility of CSCs is thought to be driven by the force of glucan chain polymerization [
15,
19].
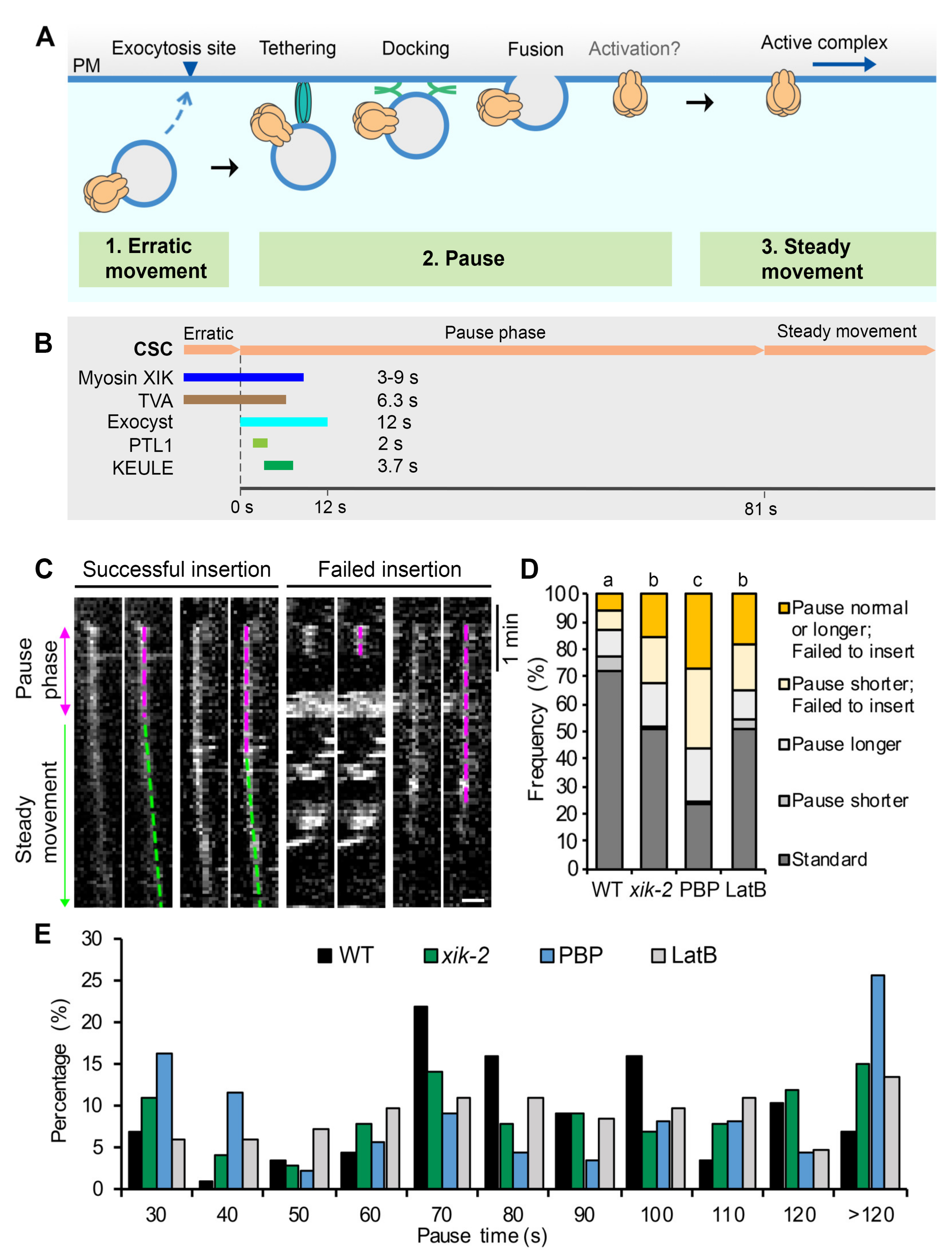
Recent advances in high spatiotemporal resolution live-cell imaging and utilization of functional, fluorescent protein-tagged CESAs have allowed CSC trafficking to emerge as a powerful model for studying exocytosis in plant cells [
15,
18,
23,
24,
25]. In particular, single-particle tracking has illuminated specific steps in the exocytosis process based on the size, unique spatial localization, and motility patterns of CSCs on different endomembrane compartments [
15,
18,
23,
24]. For example, putative CSC delivery vesicles arrive in the cortical cytoplasm near the PM, display erratic local movement for several seconds, become spatially restricted in their movement and maintain a static position for 60–90 s, and then undergo steady and linear movement as catalytically active CESA complexes (see [
18,
23,
25] and
Figure 1A). The pause phase is hypothesized to encompass tethering, docking, and fusion of CSC vesicles, possibly followed by the activation of CESAs before they synthesize cellulose and show linear motility.
Through genetic studies as well as fluorescent tagging and colocalization analyses, several proteins that transiently associate with CSC particles during the pause phase and may play a role in the efficient delivery of CSCs to the PM have been identified (
Figure 1B). Some are exocytosis-related machinery proteins, including the exocyst tethering complex which coappears with CSCs at the beginning of the pause phase for ~12 s, suggesting that vesicle tethering immediately follows the arrival of CSC compartments at the PM [
25,
26]. The plant-specific protein PATROL1 (PTL1), a homolog of Munc13 proteins in animals with a role in SNARE complex assembly, colocalizes with CSCs for 1–2 s at the pause phase and often appears 1–2 s after the appearance of exocyst subunits [
26]. KEULE, a plant homolog of Sec1/Munc18 proteins implicated in regulating SNARE function, also arrives at the beginning of the pause phase, has an average lifetime of 3.7 s, and is likely to play a role in docking and/or fusion of CSC vesicles [
27]. The actin-based motor, Myosin XIK, coappears with cortical CSC compartments and remains present at the beginning of the pause phase for 3–9 s before disappearing [
23,
25]. Finally, a new plant-specific protein, TRANVIA (TVA), arrives at the PM with CESA delivery compartments and remains associated with CSCs during the pause phase for an average of 6.3 s, and likely plays a role in CSC delivery [
24]. The tightly regulated appearance and timing of these molecular players during the initial vesicle tethering phase suggest that the pause phase is critical for complex molecular interactions that ensure successful and efficient CSC delivery to the PM. Despite the identification of several candidate proteins, the precise molecular mechanisms and sequence of events that occur during the pause phase and the details of regulation of tethering, docking, and fusion remain to be fully elucidated.
The actin cytoskeleton and myosin motors are key contributors to long distance intracellular transport in plant cells [
28,
29,
30,
31,
32]. The global distribution of CSC-containing Golgi in the cortical cytoplasm relies on an intact actin cytoskeleton network and genetic or pharmacological disruption of actin reduced the exocytosis rate of CSCs at the PM [
18,
33]. Within the myosin XI family, the XIK isoform is the major isoform responsible for organelle transport and motility in somatic cells [
31,
34]. Using CSC trafficking as a model system, we provided evidence that myosin XI participates in vesicle exocytosis near the PM and this process is likely mediated by interactions with the exocyst complex [
23,
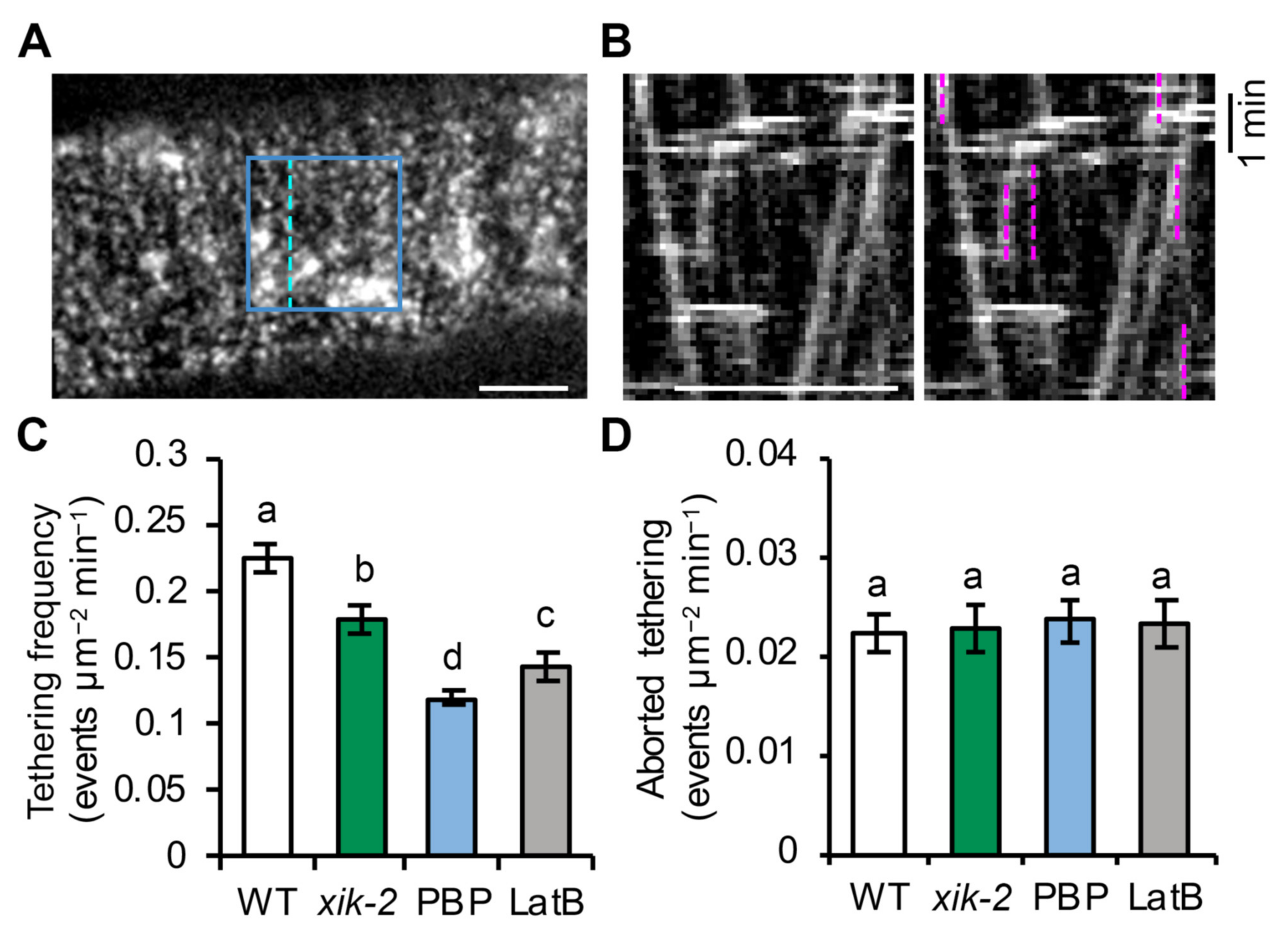
25]. Disruption of myosin XI activity, either genetically or with inhibitor treatment, results in reduced rate of overall delivery of CSCs into the PM, increased failure of late exocytosis events, and altered CSC lifetime during the pause phase [
23,
25]. Myosin XIK directly interacts with several exocyst subunits and is functionally associated with the exocyst complex at CSC delivery sites [
25]. Inhibition of myosin XIK activity results in reduced localization and a shorter lifetime of exocyst complex subunits at the PM during CSC delivery [
25]. These results indicate a role for myosin XI in the initial vesicle tethering stage; however, the exact mechanism and which sub-steps require the function of myosin remain unclear.
Despite evidence that both the actin and microtubule cytoskeletons are implicated in CSC secretion at the PM [
16,
35,
36], the exact roles they play during exocytosis remain to be fully elucidated. Here, we develop a quantitative imaging approach to directly measure the pause or tethering frequency of CSC compartments at the PM and our results confirm that actin and myosin XI are involved in the initial tethering of vesicles during CSC exocytosis. In addition, treatments with the microtubule inhibitor, oryzalin, discount a primary role for cortical microtubules in vesicle tethering and allow us to revise and expand current models for the role of cortical cytoskeleton in the last steps of CSC secretion.
3. Discussion
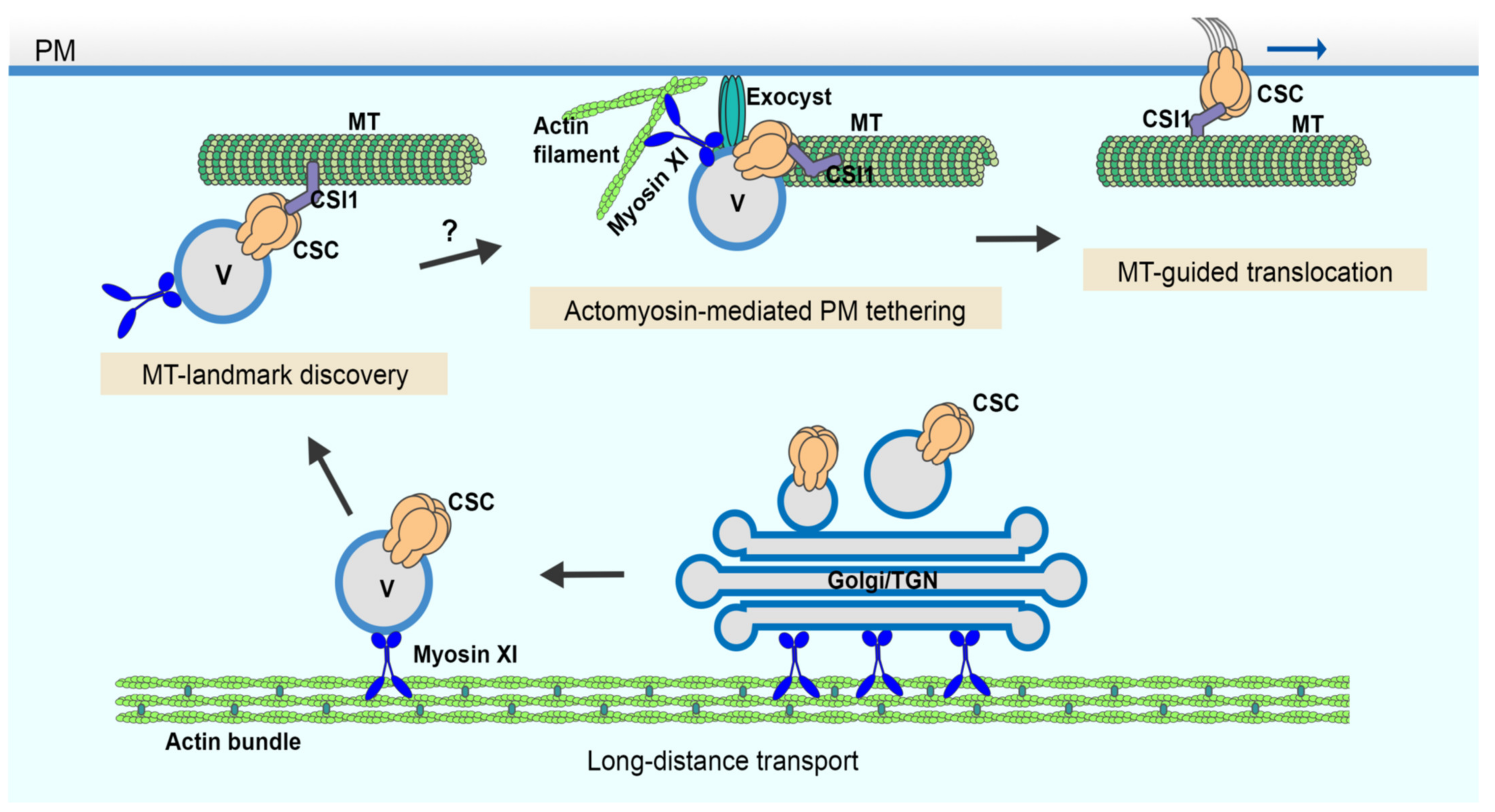
Recent advances using single-particle tracking of CSC delivery events at the PM along with genetic and small molecule inhibitor approaches reveal several evolutionarily-conserved and plant-unique players as well as specific molecular steps during late stages of the secretory processes. In this study, we developed a quantitative image analysis approach to measure vesicle tethering frequency at the PM using single-particle CSC delivery as a surrogate for exocytic events and provide direct evidence that actin and myosin XI are required for the initial vesicle tethering step during exocytosis. Further, by quantitatively assessing the late stages of CSC secretion in oryzalin-treated cells, it becomes obvious that cortical microtubules only play a minor role in vesicle tethering, docking, or insertion even though they serve as a landmark for the beginning of the pause phase (
Figure 5).
The evidence that myosin and actin play a key role during the final stages of CSC delivery is expanding rapidly. The reduced vesicle tethering rate observed here for
myosin xik as well as actin- and myosin-inhibitor treated cells is consistent with previous findings of a reduced rate of CSC delivery in
myosin xi,
actin2/7, and upon acute actin- and myosin-inhibitor treatment [
23,
33]. Moreover, abnormal accumulation of CESA compartments in the cortical cytoplasm adjacent to the PM was observed when actin or myosin function are perturbed, and is a typical phenotype of cells defective in vesicle tethering and/or fusion [
23,
25]. The accumulation of cortical CESA vesicles in actin- or myosin-deficient cells further indicates that the reduced vesicle tethering frequency is unlikely caused by a reduction in cytoplasmic streaming, which would prevent the access or delivery of vesicles to the cell cortex prior to tethering at the PM. Moreover, the timeline for arrival and disappearance of proteins at the exocytosis site can be indicative of their function during exocytosis. Previous work demonstrated that myosin XIK appearance at the CSC tethering site overlaps with the exocyst complex during the first 3–9 s of the pause phase, further supporting a role for myosin in the initial vesicle tethering step [
25]. Critically, both the lifetime and abundance of stationary exocyst complex subunits depend upon myosin XI and actin function [
25]. How myosin XI and actin coordinate with the exocyst complex and other potential players to mediate vesicle tethering requires additional investigation; however, actomyosin function could be required for the efficient recruitment and stabilization of exocyst complex to the vesicle tethering site, as suggested previously [
25].
Our latest results confirm a role for actin and myosin XI in the vesicle tethering step of exocytosis; nevertheless, we cannot exclude the possibility that actin or myosin XI are also involved in subsequent steps of exocytosis such as docking or membrane fusion. Myosin V, the closest homolog of myosin XI in animal systems, interacts with SNARE proteins and assists vesicle docking at the PM [
39]. The timing of association of myosin XIK with CSCs during the pause phase appears to partially overlap with that of PTL1 and KEULE, two proteins that are implicated in SNARE function and play a role in CSC vesicle docking and/or fusion [
26,
27]. PTL1 and KEULE show transient colocalization with CSCs during the early pause phase for only 2–4 s. The newly discovered TVA protein also colocalizes with CSC secretory vesicles and has a similar behavior to myosin XIK; it arrives with the cortical vesicle during the erratic phase and remains associated with the pause phase for ~6 s before departing [
24]. These findings suggest that TVA is another regulator of vesicle tethering and may indeed cooperate with myosins or exocyst during this key step in secretion. Interestingly, fluorescently labeled TVA moves away from the CSC tethering site as discrete particles after insertion of CSCs at the PM, resembling a kiss-and-run vesicle fusion event [
24], indicating that the molecular processes choreographing CSC secretion during the early pause phase are more complex and may require distinct mechanisms when compared with conventional exocytosis. Actin and myosin have been shown to regulate fusion pore dynamics during membrane fusion including kiss-and-run fusion events in animal exocrine cells [
40]. Further studies to dissect the role of the actin cytoskeleton and myosin XI in the docking and fusion processes as well as identification of new molecular players will help uncover the regulation of exocytosis in plant cells as well as advance our understanding of plant growth and development in general.
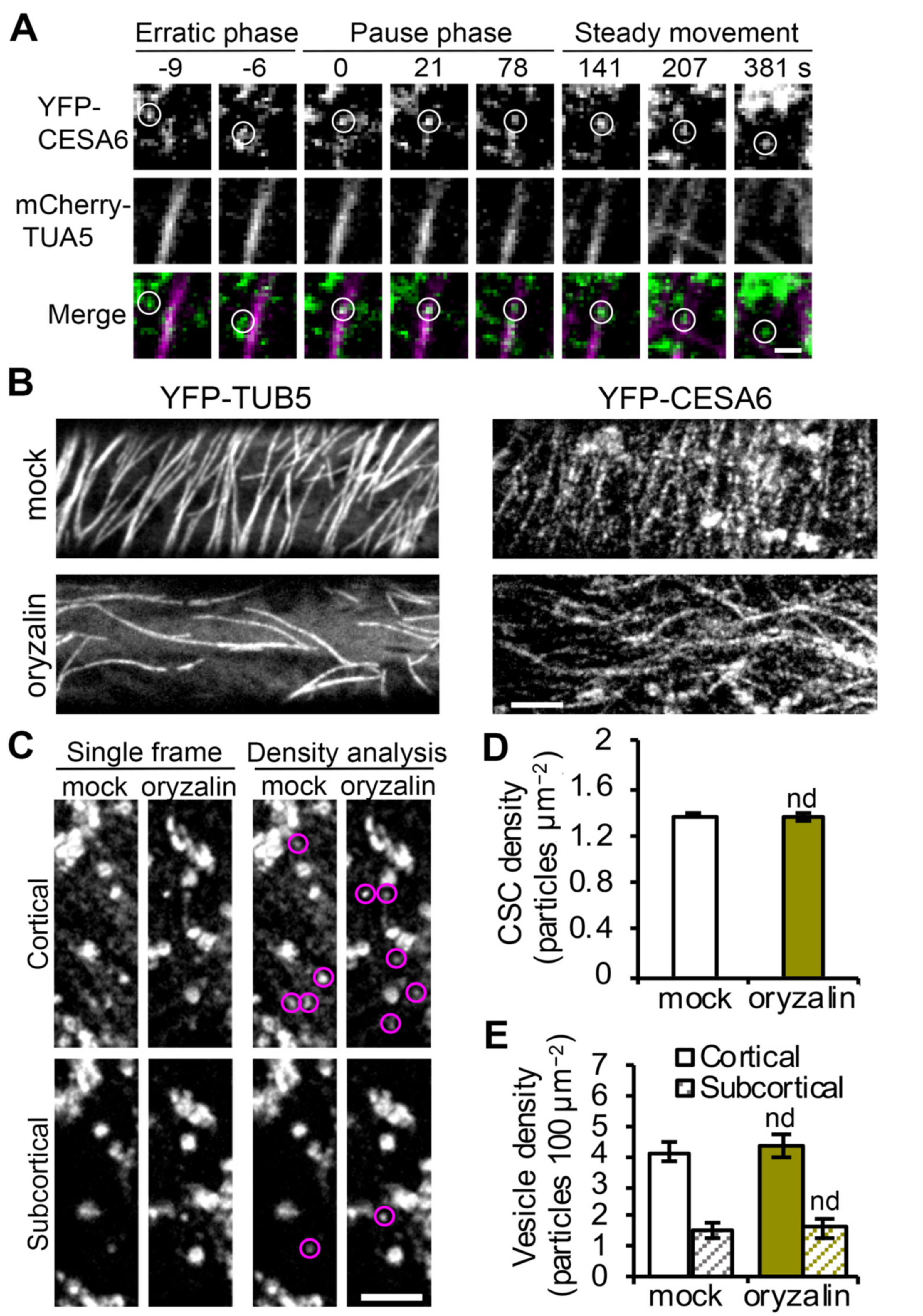
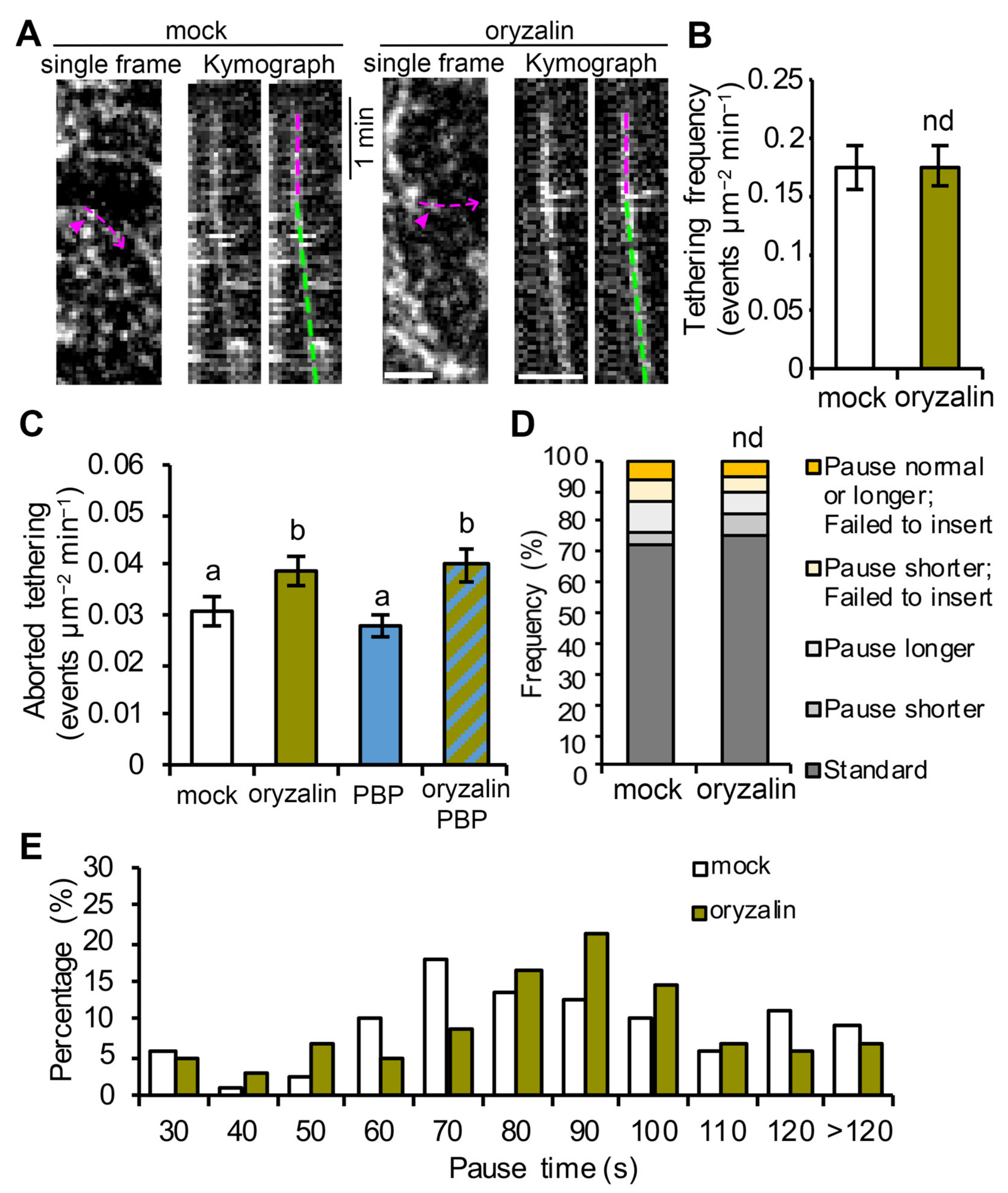
Cortical microtubules are key players in plant cell wall biosynthesis and are prominently featured in most models describing the final stages of CSC delivery to the PM [
15,
35,
41]. These models are supported by observations that 80–90% of CSCs pause on cortical microtubules prior to successful PM insertion events [
18,
21,
23]. However, the targeting of CSC compartments to cortical microtubules during PM insertion seems not to be a rate-limiting step, because other data show that depletion of microtubules with oryzalin or genetic mutation of the linker protein CSI1 does not affect the overall CSC delivery rate measured by FRAP [
18,
21,
26]. In this study, we performed CSC tethering analysis as well as single-particle CSC insertion assays to investigate the role of cortical microtubules at high spatiotemporal resolution and test directly whether cortical microtubules play a role, perhaps in coordination with actin and myosin, in facilitating efficient vesicle tethering and docking during CSC insertion. Our results showed that depletion of cortical microtubules with oryzalin did not significantly affect either the abundance of cortical CSC vesicles or the subsequent PM tethering, docking, or fusion steps during CSC exocytosis. Nevertheless, cortical microtubules may play a minor role in maintaining a stable PM tethering complex during CSC exocytosis, because we observed a 20% increase in aborted tethering events in oryzalin-treated cells. Therefore, we conclude that the targeting of CSCs to cortical microtubules is not an essential step for CSC insertion at the PM nor is it necessary for actomyosin-mediated tethering, docking and fusion steps during secretion. Perhaps the capture of CSCs on cortical microtubules simply ensures that successfully inserted CSCs are ready and able to translocate along cortical microtubules and assures that the trajectory of cellulose microfibrils in the wall is dictated by the orientation of cortical microtubule arrays (
Figure 5). Mechanistically, this could either be a redundant step or a failsafe to ensure that orientated plant cell wall deposition is primed to follow an actomyosin-dependent delivery event. We propose a revised model that PM tethering and exocytosis processes of CSC vesicles are mainly mediated through cortical actomyosin-dependent function, whereas cortical microtubules serve as a landmark to position CSC insertion sites at specific locations on the PM (
Figure 5). Although the capture on microtubules and the PM tethering of CSCs occurs roughly at the same time, the exact order of the two molecular events remains to be resolved. Quantitative, high-resolution colocalization assays with markers of microtubules, exocyst, or myosin XI as well as genetic studies with mutants of these components are required to address this question.
The involvement of both microtubule and actin cytoskeletons at PM-associated secretion sites supports prior evidence that cortical microtubules and actin filaments are often colocalized in rapid elongating cells and their organization and dynamics are interdependent [
42]. In particular, actin filament “pausing” events were frequently observed in the proximity of cortical microtubules in untreated cells and, during recovery from LatB treatments, short actin filament fragments coaligned and translocated along microtubules. It is feasible that such colocalization sites represent microtubule-defined exocytosis sites; although, it remains unclear what upstream signaling events determine their location or what molecular cues recruit secretory machinery proteins as well as microtubule- and actin-related cytoskeletal elements to these secretion sites. The function of microtubules as CSC secretion landmarks is likely to be associated with the linker protein CSI1; however, the role of CSI1 in CSC secretion needs to be further investigated. One study suggests that CSI1 localized to insertion sites many seconds prior to the arrival of CSCs [
26]; however, another report shows that only 23% of the newly-delivered CSC particles co-associate with CSI1 fluorescent puncta and the frequency of CSC insertion on cortical microtubule sites is unaltered in
csi1/pom2 mutants [
21].
At a broader level, our findings indicate an evolutionarily-conserved mechanism is shared among diverse eukaryotes and demonstrate that the late stages of exocytosis are mainly dependent on the actin cytoskeleton and myosin motors rather than microtubules.
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
An
Arabidopsis thaliana Col-0 line expressing YFP-CESA6 in the homozygous
prc1-1/cesa6 background [
19] was kindly provided by Ying Gu (Pennsylvania State University). Homozygous
myosin xik-2 and wild-type siblings expressing YFP-CESA6
prc1-1 were recovered from an F3 population resulting from the cross between a
myosin xi1,
xi2,
xik triple knockout (
xi3KO) mutant and the YFP-CESA6
prc1-1 line as described previously [
23,
25]. Transgenic plants expressing YFP-TUB5 were described previously [
38]. The YFP-CESA6 mCherry-TUA5 co-expression line was kindly provided by David W. Ehrhardt (Carnegie Institute for Science).
Arabidopsis seed was surface sterilized and stratified at 4 °C for 3 d on half-strength Murashige and Skoog (MS) medium supplemented with 0.8% agar. For light growth, plants were grown under a light intensity of 120–140 µmol m−2 s−1 under long-day conditions (16 h light/8 h dark) at 21 °C. For dark growth, plates were exposed to light for 4 h and then placed vertically and kept at 21 °C for 3 d in continuous darkness.
4.2. Live-Cell Imaging
Epidermal cells from the apical region of 3-day-old dark-grown hypocotyls were imaged by spinning-disk confocal microscopy (SDCM). Image acquisition was performed using a Yokogawa scanning unit (CSU-X1-A1; Hamamatsu Photonics, Hamamatsu, Japan) mounted on an Olympus IX-83 microscope, equipped with a 100× 1.45 numerical aperture (NA) UPlanSApo oil objective (Olympus America Inc., Waltham, MA, USA) and an Andor iXon Ultra 897BV EMCCD camera (Andor Technology, Concord, MA, USA). YFP fluorescence was excited with a 514 nm laser line and emission collected through a 542/27 nm filter. For cortical and subcortical YFP-CESA6 imaging, z-series at 0.2 µm step sizes plus time lapse with 2 s intervals for 10 frames were collected.
4.3. Image Processing and Quantitative Analysis
Image processing and analysis were performed with Fiji Is Just ImageJ [
43].
A high spatiotemporal resolution single-particle CSC insertion assay was performed as described previously [
23]. Time series collected by SDCM at 3 s intervals for 10 min were used for this analysis. Only CSC particles that showed de novo appearance at the plane of the PM followed by a pause phase of more than 5 frames (>15 s) were considered to be new insertion events. The presence and duration of the pause phase was determined by analysis of kymographs. A line was drawn along the trajectory of a newly inserted CSC particle and a kymograph was generated with the Multi Kymograph function in FIJI. A straight vertical line on the kymograph was considered to be a pause event. The duration of the particle pause phase was determined by fitting a straight line along the path of continuous movement and another line along the pause phase on the kymograph. The intersection of the two lines was defined as the end of the pause phase. For quantification, 10 random insertion events were analyzed in each cell and a total of 10–12 cells were measured from at least 5 seedlings per genotype or treatment.
To analyze CSC tethering frequency, the same SDCM time series used for the single-particle insertion assay collected at 3 s intervals for 10 min were used. To estimate a tethering frequency (number of events per unit area and time), a region of interest (ROI) with a size of 60 × 60 pixels2 box (63.68 µm2) at the PM was chosen and all tethering events were measured within the ROI during a 6 min time span. Kymograph analysis was used to quantify tethering events. To aid in detection of tethering events on kymographs, 2-frame averaging was applied to the time series with the Group Z Project function in FIJI using the average intensity method. The time series were pre-rotated to make sure that most of the CESA trajectories were fitting vertical lines which is the same orientation that all the kymographs were generated. To create kymographs that cover all of the particle trajectories in the ROI, the Reslice function in FIJI was used and “start at left” was chosen. All vertical lines on kymographs that lasted for 30 s or longer were counted as vesicle tethering events regardless of whether they were successful insertions or not. The frequency of tethering was calculated as the number of tethering events per unit area divided by elapsed time. For quantification of transient/aborted tethering events, SDCM timelapse images collected at 1 s intervals for 6 min were used. The same kymograph approach described above was used to identify transient tethering events and only vertical lines that had a duration of longer than 3 s but less than 30 s were counted.
Analysis of PM-localized CSC density and cortical and subcortical CESA compartment density were performed as described previously [
23].
4.4. Drug Treatment
For short-term live cell treatments, seedlings were submerged in mock or inhibitor solution in a 24-well plate or directly on the slide and kept in the dark prior to mounting and imaging. The myosin inhibitor pentabromopseudilin (PBP; Adipogen, San Diego, CA, USA), actin polymerization inhibitor latrunculin B (LatB; Calbiochem, San Diego, CA, USA), and microtubule depolymerizing agent oryzalin (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in DMSO to generate stock solutions that were stored at −20 °C and diluted in water immediately prior to use.
4.5. Statistical Analysis
One-way ANOVA with Tukey’s post-hoc tests were performed in SPSS (version 27) to determine significance among different treatments. Two-tailed Student’s t-tests were performed in Excel 16.51. Chi-square tests were used for statistical comparison of data that did not follow parametric distributions and p values were calculated in Excel 16.51.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




