
The Osteogenesis Imperfecta Type V Mutant BRIL/IFITM5 Promotes Transcriptional Activation of MEF2, NFATc, and NR4A in Osteoblasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
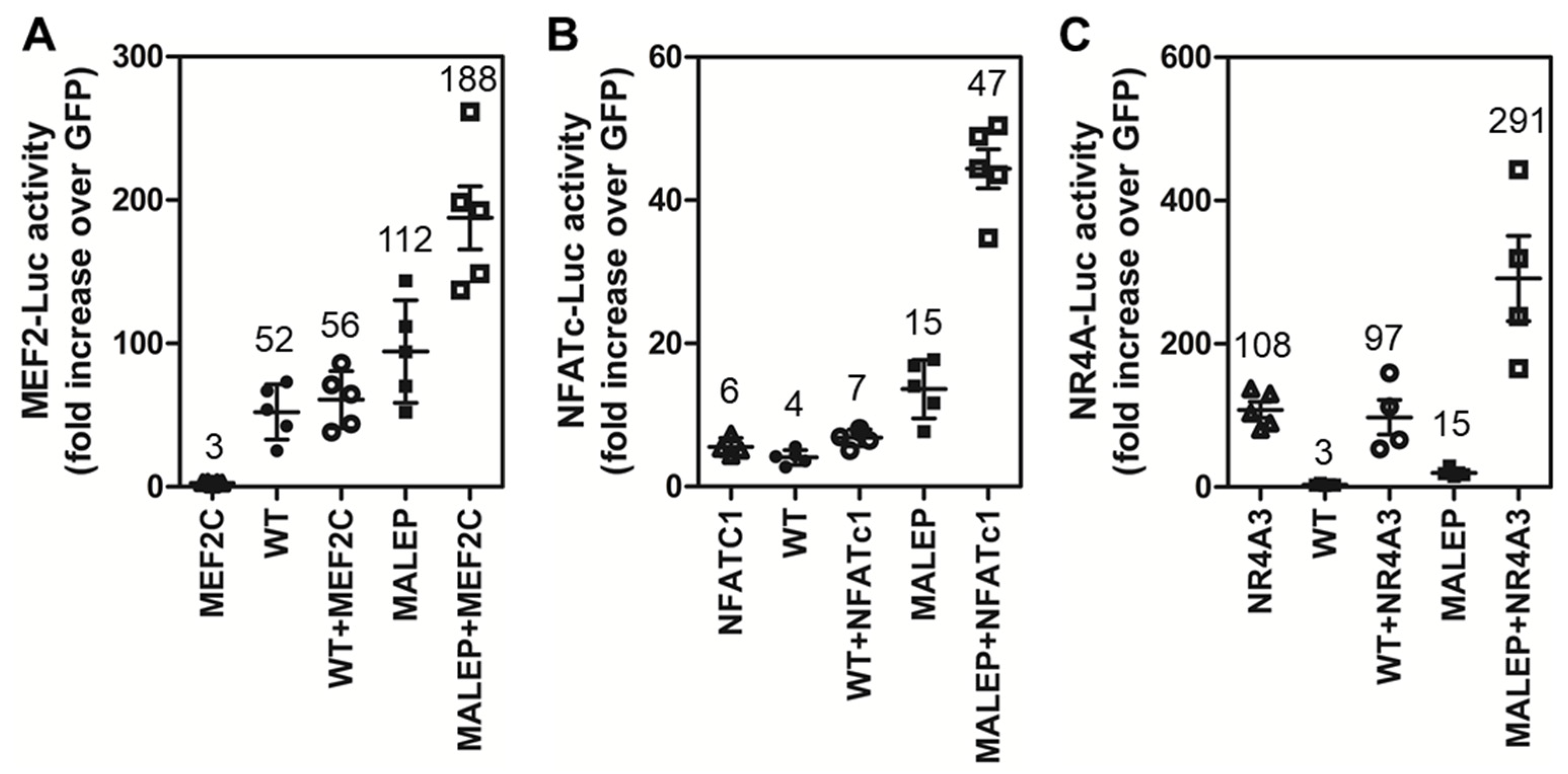
2.1. BRIL Induces Pathways Leading to Activation of the MEF2-, NFATc-, and NR4A-Luc Reporters
2.2. The Mouse and Human Wild Type (WT) and Mutant (MALEP) BRIL Activate MEF2- and NFATC-Luc Reporters in MC3T3-E1
2.3. Overexpression of BRIL Does Not Lead to Increased Transcription of the Mef and NFATc Genes
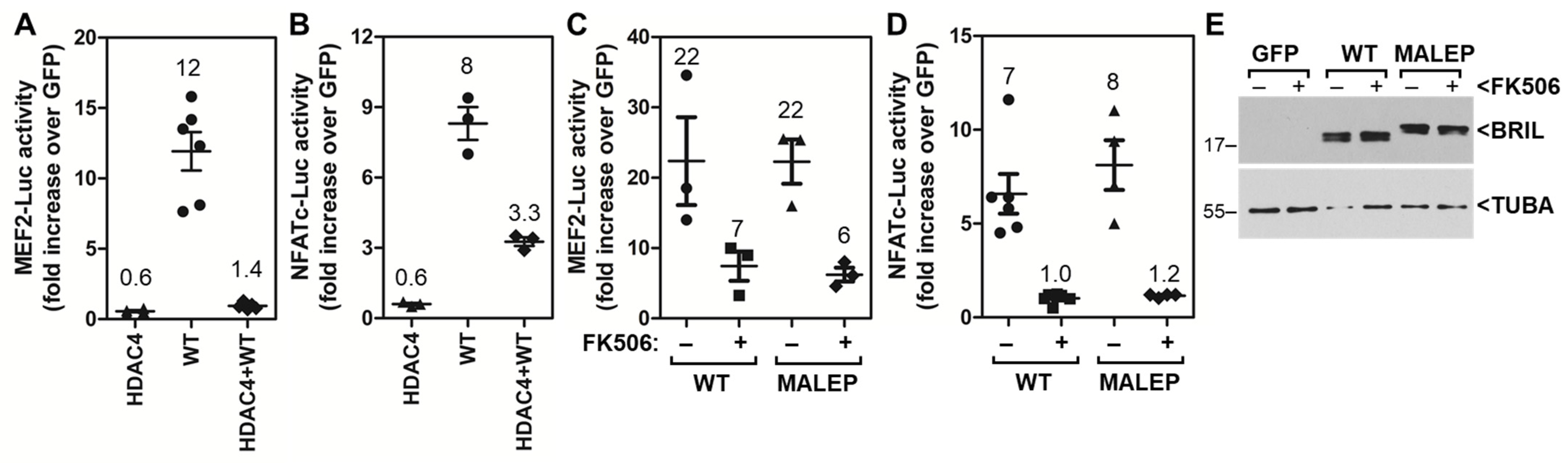
2.4. The Ability of WT and MALEP BRIL to Induce MEF2 and NFATc Is Repressed by HDAC4 and FK506
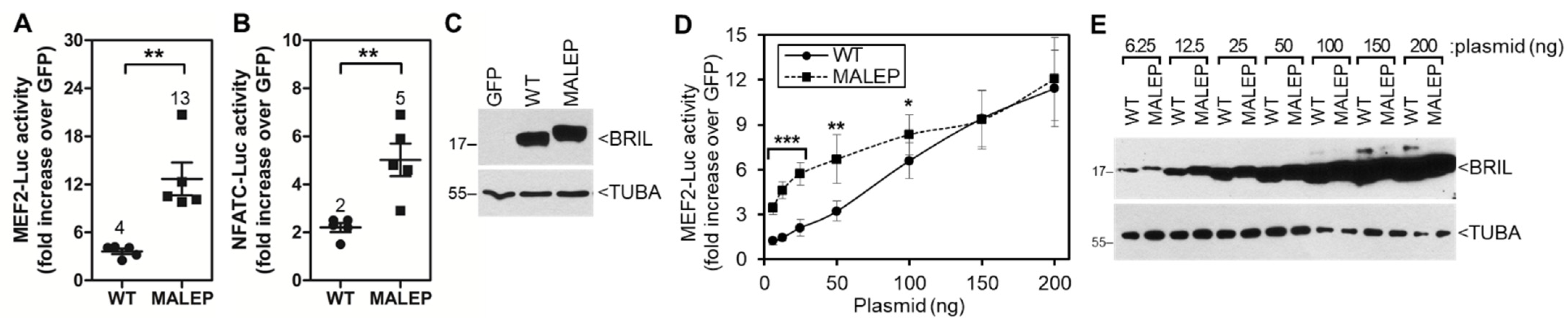
2.5. Time-Course and Dose-Response for WT and MALEP BRIL Activity in MC3T3-E1
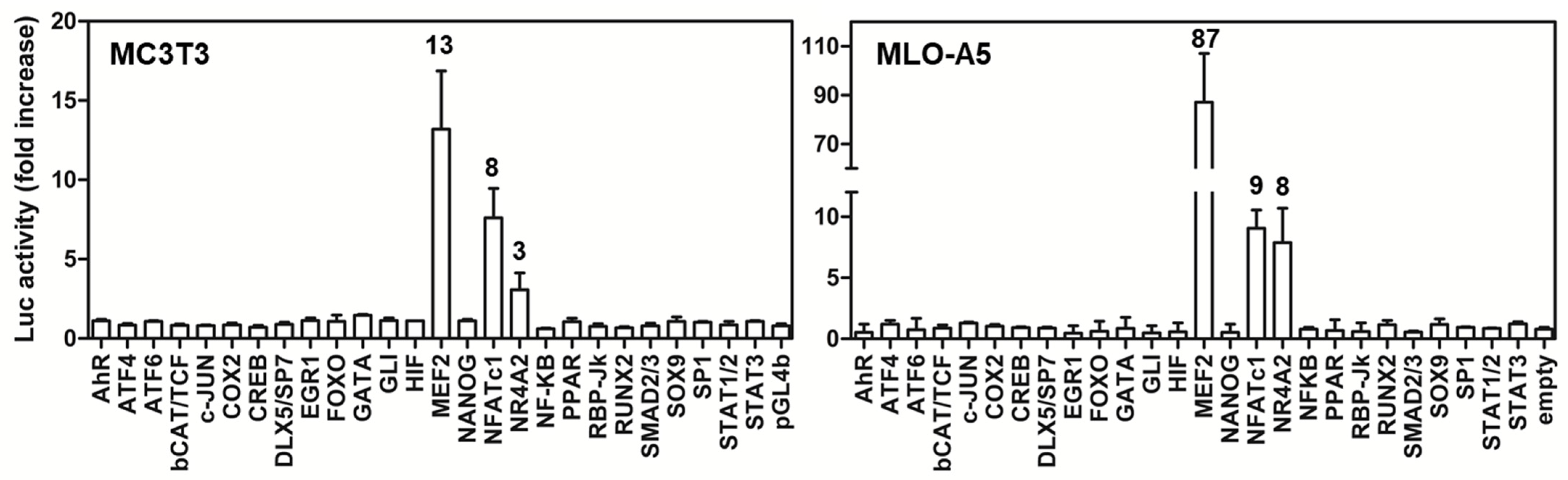
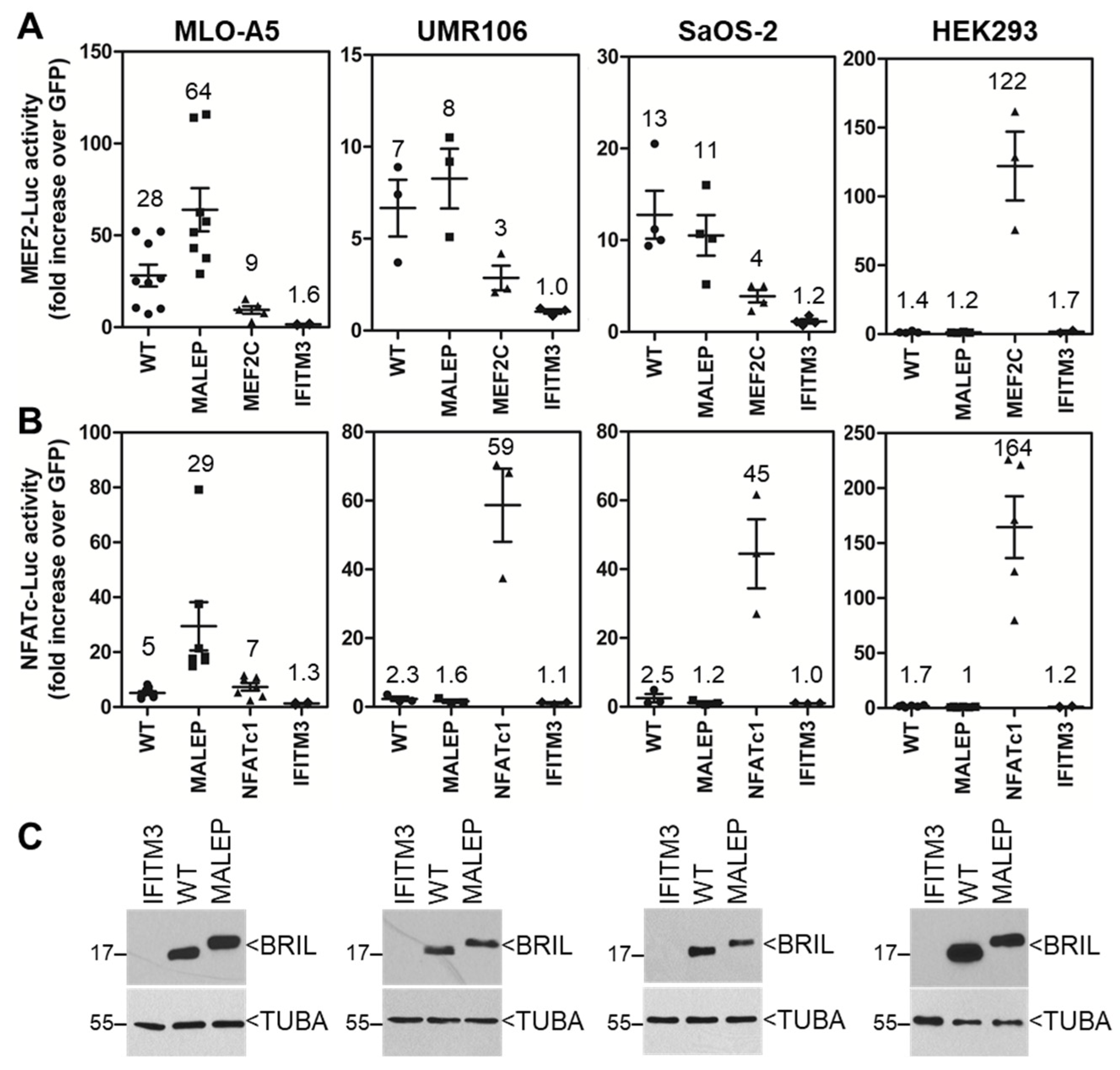
2.6. Screening of the Activity of BRIL on MEF2-Luc and NFATc-Luc in Various Cell Lines
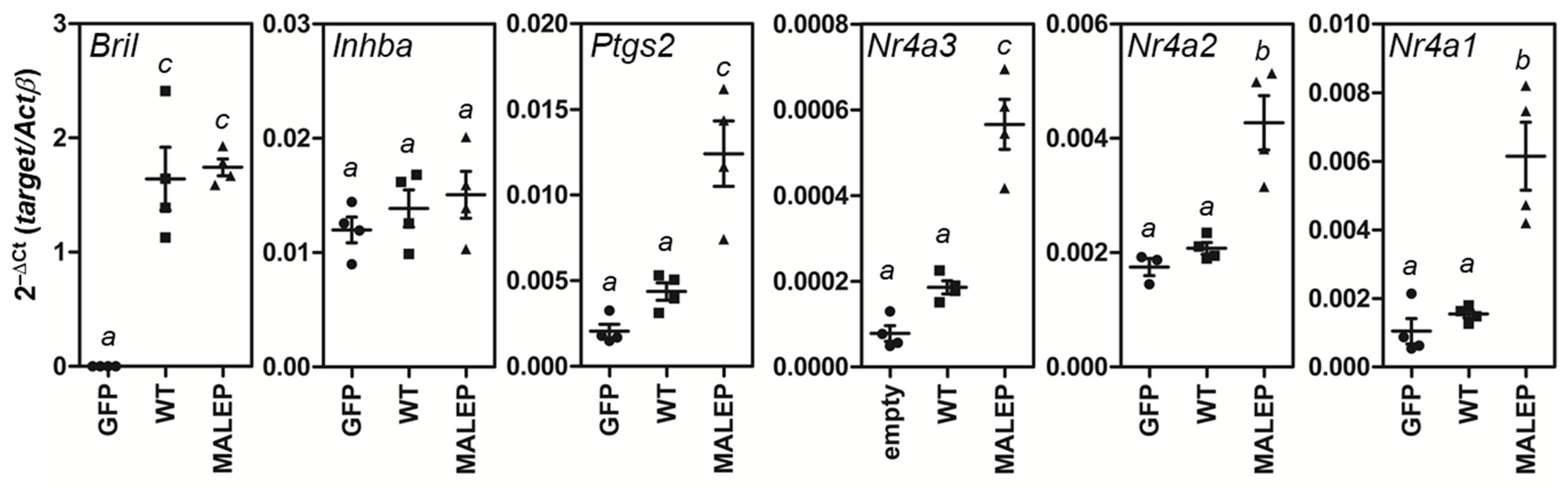
2.7. MALEP BRIL Selectively Induces Gene Expression of Nr4a Members and Ptgs2 in MLO-A5 Cells
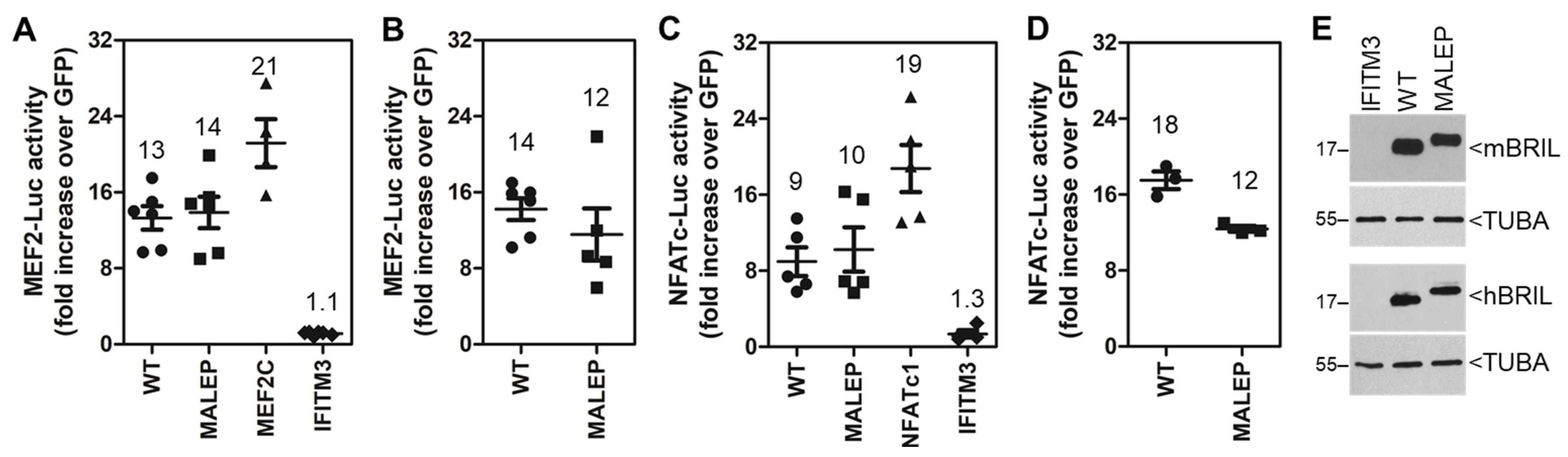
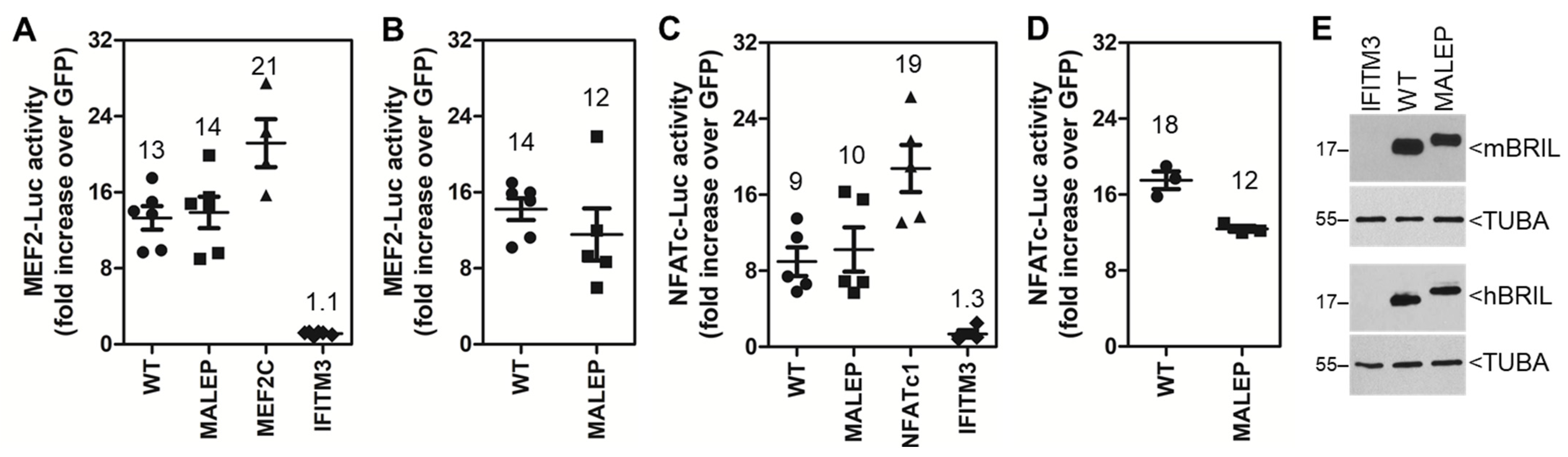
2.8. Differences in the Activity of WT and MALEP BRIL to Regulate MEF2, NFATc, and NR4A Reporters
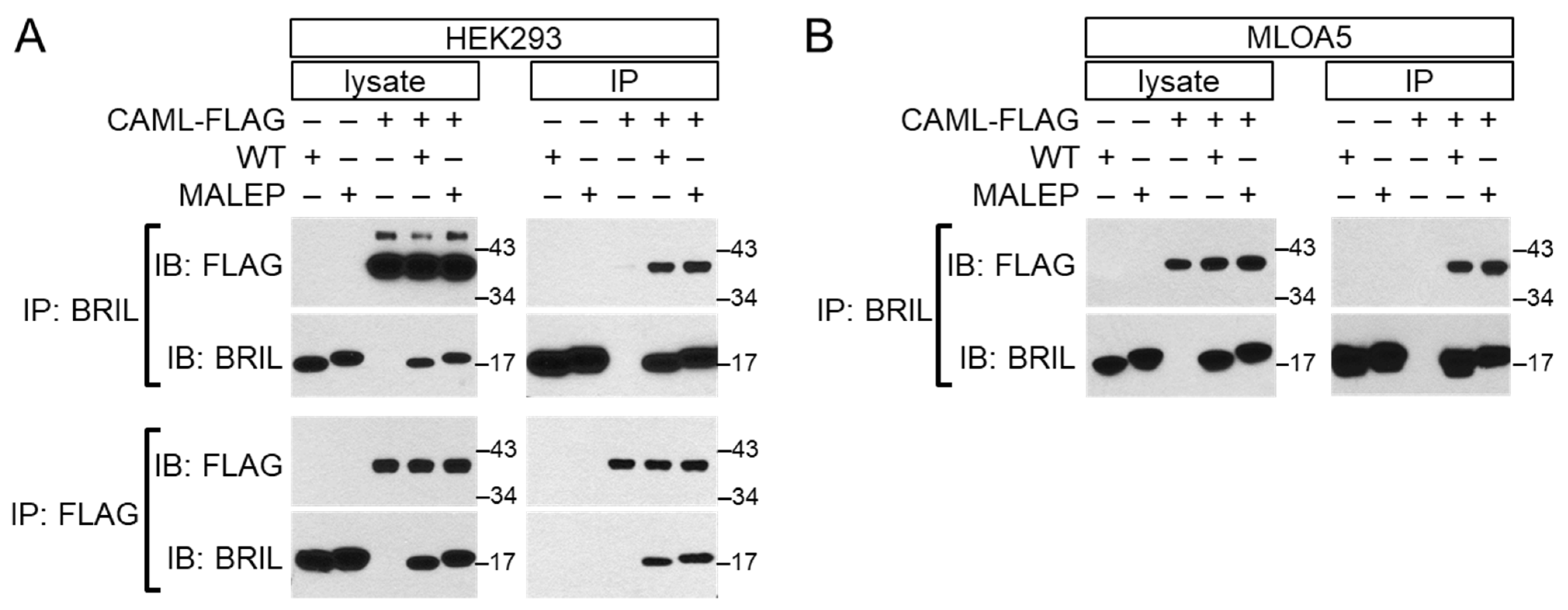
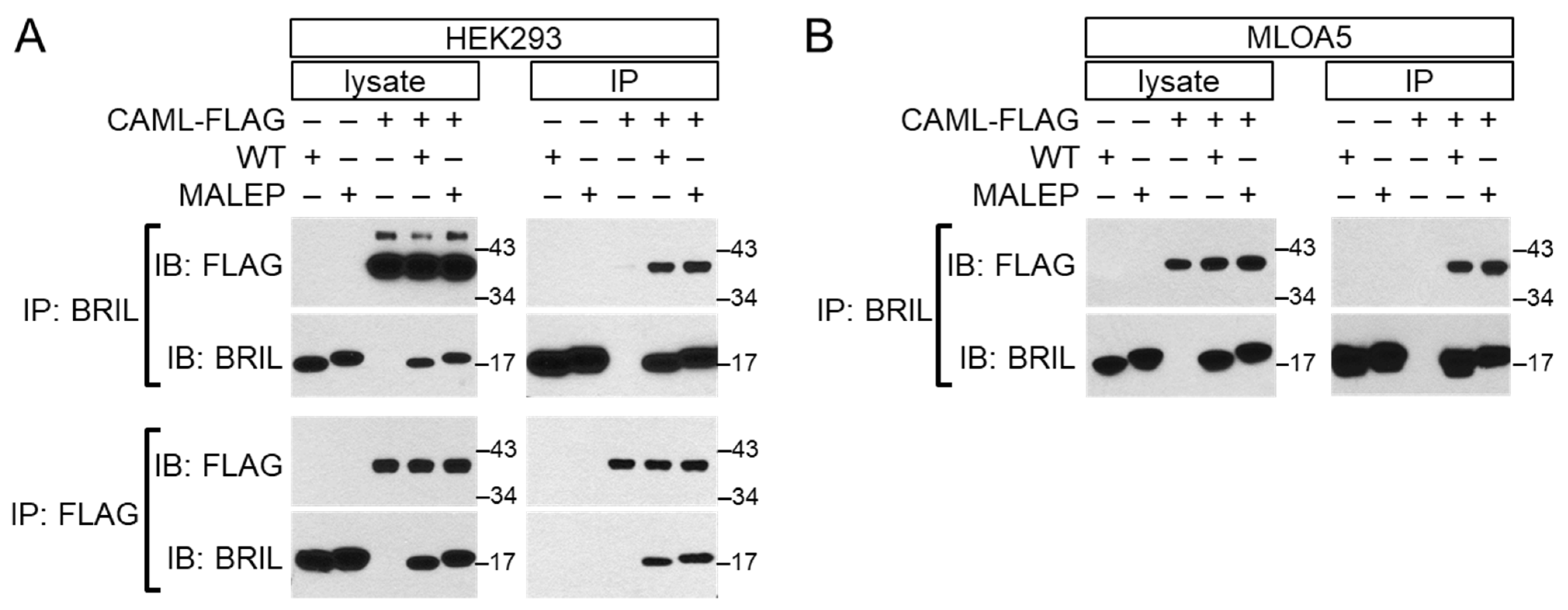
2.9. Interaction of BRIL with CAML by Yeast 2-Hybrid and Validation by Co-Immunoprecipitation
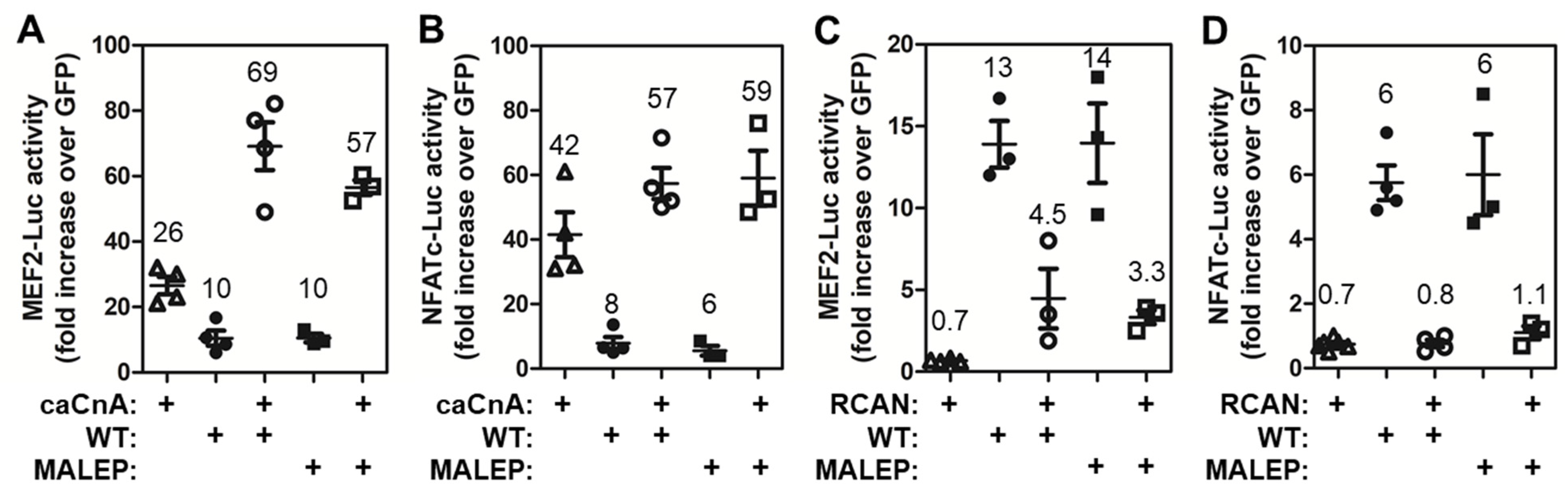
2.10. CAML Differentially Regulates the Activity of MEF2 and NFATc
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Cloning and Luciferase (Luc) Reporter- and Expression-Plasmids
4.3. Luc Assay
4.4. RNA Extraction and RT-qPCR
4.5. Protein Extraction, Western Blotting and Co-Immunoprecipitation
4.6. Yeast 2-Hybrid (Y2H) Screen
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Rauch, F.; Glorieux, F.H. Osteogenesis imperfecta. Lancet 2004, 363, 1377–1385. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Bachinger, H.P.; Bishop, N.J.; Byers, P.H.; Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 2017, 3, 17052. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Blissett, A.R. New Genes in Bone Development: What’s New in Osteogenesis Imperfecta. J. Clin. Endocrinol. Metab. 2013, 98, 3095–3103. [Google Scholar] [CrossRef] [PubMed]
- Semler, O.; Garbes, L.; Keupp, K.; Swan, D.; Zimmermann, K.; Becker, J.; Iden, S.; Wirth, B.; Eysel, P.; Koerber, F.; et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am. J. Hum. Genet. 2012, 91, 349–357. [Google Scholar] [CrossRef]
- Cho, T.J.; Lee, K.E.; Lee, S.K.; Song, S.J.; Kim, K.J.; Jeon, D.; Lee, G.; Kim, H.N.; Lee, H.R.; Eom, H.H.; et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am. J. Hum. Genet. 2012, 91, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Farber, C.R.; Reich, A.; Barnes, A.M.; Becerra, P.; Rauch, F.; Cabral, W.A.; Bae, A.; Quinlan, A.; Glorieux, F.H.; Clemens, T.L.; et al. A novel IFITM5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J. Bone Miner. Res. 2014, 29, 1402–1411. [Google Scholar] [CrossRef]
- Hoyer-Kuhn, H.; Semler, O.; Garbes, L.; Zimmermann, K.; Becker, J.; Wollnik, B.; Schoenau, E.; Netzer, C. A nonclassical IFITM5 mutation located in the coding region causes severe osteogenesis imperfecta with prenatal onset. J. Bone Miner. Res. 2014, 29, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Bhatia, N.S.; Vasanwala, R.F.; Chay, P.L.; Lim, K.B.L.; Khoo, P.C.; Schwarze, U.; Jamuar, S.S. A novel Ser40Trp variant in IFITM5 in a family with osteogenesis imperfecta and review of the literature. Clin. Dysmorphol. 2019, 28, 120–125. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, S.; Huang, X.; Pang, J.; Liu, J.; Hu, J.; Shen, X.; Tang, C.; Wang, H. Whole Exome Sequencing Analysis in Fetal Skeletal Dysplasia Detected by Ultrasonography: An Analysis of 38 Cases. Front. Genet. 2021, 12, 728544. [Google Scholar] [CrossRef]
- Makitie, R.E.; Pekkinen, M.; Morisada, N.; Kobayashi, D.; Yonezawa, Y.; Nishimura, G.; Ikegawa, S.; Makitie, O. A Novel IFITM5 Variant Associated with Phenotype of Osteoporosis with Calvarial Doughnut Lesions: A Case Report. Calcif. Tissue Int. 2021, 109, 626–632. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Y.; Huang, H. A novel variant of the IFITM5 gene within the 5′-UTR causes neonatal transverse clavicular fracture: Expanding the genetic spectrum. Mol. Genet. Genom. Med. 2020, 8, e1287. [Google Scholar] [CrossRef] [PubMed]
- Bardai, G.; Moffatt, P.; Glorieux, F.H.; Rauch, F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: Diagnostic yield and mutation spectrum. Osteoporos. Int. 2016, 27, 3607–3613. [Google Scholar] [CrossRef] [PubMed]
- Reich, A.; Bae, A.S.; Barnes, A.M.; Cabral, W.A.; Hinek, A.; Stimec, J.; Hill, S.C.; Chitayat, D.; Marini, J.C. Type V OI primary osteoblasts display increased mineralization despite decreased COL1A1 expression. J. Clin. Endocrinol. Metab. 2015, 100, E325–E332. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.R.; Lietman, C.; Grover, M.; Lu, J.T.; Nagamani, S.C.; Dawson, B.C.; Baldridge, D.M.; Bainbridge, M.N.; Cohn, D.H.; Blazo, M.; et al. Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J. Bone Miner. Res. 2013, 28, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Rauch, F.; Moffatt, P.; Cheung, M.; Roughley, P.; Lalic, L.; Lund, A.M.; Ramirez, N.; Fahiminiya, S.; Majewski, J.; Glorieux, F.H. Osteogenesis imperfecta type V: Marked phenotypic variability despite the presence of the IFITM5 c.-14C>T mutation in all patients. J. Med. Genet. 2013, 50, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, P.; Gaumond, M.H.; Salois, P.; Sellin, K.; Bessette, M.C.; Godin, E.; de Oliveira, P.T.; Atkins, G.J.; Nanci, A.; Thomas, G. Bril: A novel bone-specific modulator of mineralization. J. Bone Miner. Res. 2008, 23, 1497–1508. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Rauch, F.; Plotkin, H.; Ward, L.; Travers, R.; Roughley, P.; Lalic, L.; Glorieux, D.F.; Fassier, F.; Bishop, N.J. Type V osteogenesis imperfecta: A new form of brittle bone disease. J. Bone Miner. Res. 2000, 15, 1650–1658. [Google Scholar] [CrossRef]
- Cheung, M.S.; Glorieux, F.H.; Rauch, F. Natural history of hyperplastic callus formation in osteogenesis imperfecta type V. J. Bone Miner. Res. 2007, 22, 1181–1186. [Google Scholar] [CrossRef]
- Cheung, M.S.; Azouz, E.M.; Glorieux, F.H.; Rauch, F. Hyperplastic callus formation in osteogenesis imperfecta type V: Follow-up of three generations over ten years. Skelet. Radiol. 2008, 37, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Blouin, S.; Fratzl-Zelman, N.; Glorieux, F.H.; Roschger, P.; Klaushofer, K.; Marini, J.C.; Rauch, F. Hypermineralization and High Osteocyte Lacunar Density in Osteogenesis Imperfecta Type V Bone Indicate Exuberant Primary Bone Formation. J. Bone Miner. Res. 2017, 32, 1884–1892. [Google Scholar] [CrossRef]
- Hickford, D.; Frankenberg, S.; Shaw, G.; Renfree, M.B. Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genom. 2012, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Sallman, A.M.; Bringeland, N.; Fredriksson, R.; Schioth, H.B. The dispanins: A novel gene family of ancient origin that contains 14 human members. PLoS ONE 2012, 7, e31961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Li, M.; Yang, H.; Zhang, C. Evolutionary dynamics of the interferon-induced transmembrane gene family in vertebrates. PLoS ONE 2012, 7, e49265. [Google Scholar] [CrossRef] [PubMed]
- Patoine, A.; Gaumond, M.H.; Jaiswal, P.K.; Fassier, F.; Rauch, F.; Moffatt, P. Topological mapping of BRIL reveals a type II orientation and effects of osteogenesis imperfecta mutations on its cellular destination. J. Bone Miner. Res. 2014, 29, 2004–2016. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Li, X.; Morita, H.; Minowa, T.; Aizawa, T.; Hanagata, N.; Demura, M. Role of S-palmitoylation on IFITM5 for the interaction with FKBP11 in osteoblast cells. PLoS ONE 2013, 8, e75831. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e6. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Ono, N.; Ayturk, U.M.; Debnath, S.; Lalani, S. The Unmixing Problem: A Guide to Applying Single-Cell RNA Sequencing to Bone. J. Bone Miner. Res. 2019, 34, 1207–1219. [Google Scholar] [CrossRef]
- Matsushita, Y.; Nagata, M.; Kozloff, K.M.; Welch, J.D.; Mizuhashi, K.; Tokavanich, N.; Hallett, S.A.; Link, D.C.; Nagasawa, T.; Ono, W.; et al. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat. Commun. 2020, 11, 332. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Youlten, S.E.; Kemp, J.P.; Logan, J.G.; Ghirardello, E.J.; Sergio, C.M.; Dack, M.R.G.; Guilfoyle, S.E.; Leitch, V.D.; Butterfield, N.C.; Komla-Ebri, D.; et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 2021, 12, 2444. [Google Scholar] [CrossRef]
- Lange, U.C.; Adams, D.J.; Lee, C.; Barton, S.; Schneider, R.; Bradley, A.; Surani, M.A. Normal germ line establishment in mice carrying a deletion of the Ifitm/Fragilis gene family cluster. Mol. Cell. Biol. 2008, 28, 4688–4696. [Google Scholar] [CrossRef] [PubMed]
- Hanagata, N.; Li, X.; Morita, H.; Takemura, T.; Li, J.; Minowa, T. Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J. Bone Miner. Metab. 2011, 29, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Patoine, A.; Husseini, A.; Kasaai, B.; Gaumond, M.H.; Moffatt, P. The osteogenic cell surface marker BRIL/IFITM5 is dispensable for bone development and homeostasis in mice. PLoS ONE 2017, 12, e0184568. [Google Scholar] [CrossRef] [PubMed]
- Lietman, C.D.; Marom, R.; Munivez, E.; Bertin, T.K.; Jiang, M.M.; Chen, Y.; Dawson, B.; Weis, M.A.; Eyre, D.; Lee, B. A transgenic mouse model of OI type V supports a neomorphic mechanism of the IFITM5 mutation. J. Bone Miner. Res. 2015, 30, 489–498. [Google Scholar] [CrossRef]
- Rauch, F.; Geng, Y.; Lamplugh, L.; Hekmatnejad, B.; Gaumond, M.H.; Penney, J.; Yamanaka, Y.; Moffatt, P. Crispr-Cas9 engineered osteogenesis imperfecta type V leads to severe skeletal deformities and perinatal lethality in mice. Bone 2018, 107, 131–142. [Google Scholar] [CrossRef]
- Hanagata, N.; Takemura, T.; Kamimura, K.; Koda, T. Effect of immunosuppressants on a mouse model of osteogenesis imperfecta type V harboring a heterozygous Ifitm5 c.-14C > T mutation. Sci. Rep. 2020, 10, 21197. [Google Scholar] [CrossRef]
- Al, K.A.; Ganger, R.; Klaushofer, K.; Grill, F. Swellings over the Limbs as the Earliest Feature in a Patient with Osteogenesis Imperfecta Type V. Case Rep. Orthop. 2014, 2014, 780959. [Google Scholar]
- Salter, L.; Offiah, A.C.; Bishop, N. Elevated platelet counts in a cohort of children with moderate-severe osteogenesis imperfecta suggest that inflammation is present. Arch. Dis. Child. 2018, 103, 767–771. [Google Scholar] [CrossRef]
- Lazarus, S.; McInerney-Leo, A.M.; McKenzie, F.A.; Baynam, G.; Broley, S.; Cavan, B.V.; Munns, C.F.; Pruijs, J.E.; Sillence, D.; Terhal, P.A.; et al. The IFITM5 mutation c.-14C > T results in an elongated transcript expressed in human bone; and causes varying phenotypic severity of osteogenesis imperfecta type V. BMC Musculoskelet. Disord. 2014, 15, 107. [Google Scholar] [CrossRef]
- Sudo, H.; Kodama, H.A.; Amagai, Y.; Yamamoto, S.; Kasai, S. In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J. Cell Biol. 1983, 96, 191–198. [Google Scholar] [CrossRef]
- Kato, Y.; Boskey, A.; Spevak, L.; Dallas, M.; Hori, M.; Bonewald, L.F. Establishment of an osteoid preosteocyte-like cell MLO-A5 that spontaneously mineralizes in culture. J. Bone Miner. Res. 2001, 16, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Brenner, R.E.; Vetter, U.; Nerlich, A.; Worsdorfer, O.; Teller, W.M.; Muller, P.K. Biochemical analysis of callus tissue in osteogenesis imperfecta type IV. Evidence for transient overmodification in collagen types I and III. J. Clin. Investig. 1989, 84, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Aronson, J.; McAlister, W.H.; Weinstein, R.S.; Wenkert, D.; Clements, K.L.; Gottesman, G.S.; Madson, K.L.; Stolina, M.; Bijanki, V.N.; et al. Coalescing expansile skeletal disease: Delineation of an extraordinary osteopathy involving the IFITM5 mutation of osteogenesis imperfecta type V. Bone 2021, 145, 115835. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.B.; Hu, J.; Zhang, J.; Yang, Z.; Wang, O.; Jiang, Y.; Xia, W.B.; Xing, X.P.; Yu, W.; Li, M. Specific Characteristic of Hyperplastic Callus in a Larger Cohort of Osteogenesis Imperfecta Type V. Calcif. Tissue Int. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, S.E.; Nicol, L.E.; Gamayo, A.C.; Raney, E.M. An Unusual Presentation of Osteogenesis Imperfecta: A Case Report. JBJS Case Connect. 2021, 11, e21. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Huo, Y.; Li, J. Case Report: Hyperplastic Callus of the Femur Mimicking Osteosarcoma in Osteogenesis Imperfecta Type V. Front. Endocrinol. 2021, 12, 622674. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.; Kanza, R.E.; Barabas, D.; Lessard, M.; Berube, M. Non-traumatic hypertrophic callus of the fibula mimicking osteosarcoma in osteogenesis imperfecta type V: A case report. Skelet. Radiol. 2014, 43, 1333–1336. [Google Scholar] [CrossRef]
- Gregoire, S.; Tremblay, A.M.; Xiao, L.; Yang, Q.; Ma, K.; Nie, J.; Mao, Z.; Wu, Z.; Giguere, V.; Yang, X.J. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J. Biol. Chem. 2006, 281, 4423–4433. [Google Scholar] [CrossRef]
- Duran, J.; Lagos, D.; Pavez, M.; Troncoso, M.F.; Ramos, S.; Barrientos, G.; Ibarra, C.; Lavandero, S.; Estrada, M. Ca2+/Calmodulin-Dependent Protein Kinase II and Androgen Signaling Pathways Modulate MEF2 Activity in Testosterone-Induced Cardiac Myocyte Hypertrophy. Front. Pharmacol. 2017, 8, 604. [Google Scholar] [CrossRef]
- Zhang, M.; Hagenmueller, M.; Riffel, J.H.; Kreusser, M.M.; Bernhold, E.; Fan, J.; Katus, H.A.; Backs, J.; Hardt, S.E. Calcium/calmodulin-dependent protein kinase II couples Wnt signaling with histone deacetylase 4 and mediates dishevelled-induced cardiomyopathy. Hypertension 2015, 65, 335–344. [Google Scholar] [CrossRef]
- Li, S.; Keung, W.; Cheng, H.; Li, R.A. Structural and Mechanistic Bases of Nuclear Calcium Signaling in Human Pluripotent Stem Cell-Derived Ventricular Cardiomyocytes. Stem Cells Int. 2019, 2019, 8765752. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Aponte Paris, S.; Thakur, H.; Kapiloff, M.S.; Dodge-Kafka, K.L. Muscle A-kinase-anchoring protein-beta-bound calcineurin toggles active and repressive transcriptional complexes of myocyte enhancer factor 2D. J. Biol. Chem. 2019, 294, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.S.; Stephens, S.R.; Hobbs, C.; Hutmacher, D.W.; Bacic-Welsh, D.; Woodruff, M.A.; Morrison, N.A. Myocyte enhancer factor 2c, an osteoblast transcription factor identified by dimethyl sulfoxide (DMSO)-enhanced mineralization. J. Biol. Chem. 2011, 286, 30071–30086. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.W.; Tan, Z.; To, M.K.T.; Chan, D. Regulation and Role of Transcription Factors in Osteogenesis. Int. J. Mol. Sci. 2021, 22, 5445. [Google Scholar] [CrossRef]
- Kawane, T.; Komori, H.; Liu, W.; Moriishi, T.; Miyazaki, T.; Mori, M.; Matsuo, Y.; Takada, Y.; Izumi, S.; Jiang, Q.; et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J. Bone Miner. Res. 2014, 29, 1960–1969. [Google Scholar] [CrossRef]
- Kramer, I.; Baertschi, S.; Halleux, C.; Keller, H.; Kneissel, M. Mef2c deletion in osteocytes results in increased bone mass. J. Bone Miner. Res. 2012, 27, 360–373. [Google Scholar] [CrossRef]
- Arnold, M.A.; Kim, Y.; Czubryt, M.P.; Phan, D.; McAnally, J.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 2007, 12, 377–389. [Google Scholar] [CrossRef]
- Kang, J.Y.; Kang, N.; Yang, Y.M.; Hong, J.H.; Shin, D.M. The Role of Ca2+-NFATc1 Signaling and Its Modulation on Osteoclastogenesis. Int. J. Mol. Sci. 2020, 21, 3646. [Google Scholar] [CrossRef]
- Winslow, M.M.; Pan, M.; Starbuck, M.; Gallo, E.M.; Deng, L.; Karsenty, G.; Crabtree, G.R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell 2006, 10, 771–782. [Google Scholar] [CrossRef]
- Sesler, C.L.; Zayzafoon, M. NFAT signaling in osteoblasts regulates the hematopoietic niche in the bone microenvironment. Clin. Dev. Immunol. 2013, 2013, 107321. [Google Scholar] [CrossRef]
- Lee, H.L.; Bae, O.Y.; Baek, K.H.; Kwon, A.; Hwang, H.R.; Qadir, A.S.; Park, H.J.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. High extracellular calcium-induced NFATc3 regulates the expression of receptor activator of NF-kappaB ligand in osteoblasts. Bone 2011, 49, 242–249. [Google Scholar] [CrossRef]
- Yeo, H.; Beck, L.H.; Thompson, S.R.; Farach-Carson, M.C.; McDonald, J.M.; Clemens, T.L.; Zayzafoon, M. Conditional disruption of calcineurin B1 in osteoblasts increases bone formation and reduces bone resorption. J. Biol. Chem. 2007, 282, 35318–35327. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Matsui, Y.; Asagiri, M.; Kodama, T.; de Crombrugghe, B.; Nakashima, K.; Takayanagi, H. NFAT and Osterix cooperatively regulate bone formation. Nat. Med. 2005, 11, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Choo, M.K.; Yeo, H.; Zayzafoon, M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone 2009, 45, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Shing, J.C.; Bram, R.J. Yet another hump for CAML: Support of cell survival independent of tail-anchored protein insertion. Cell Death Dis. 2017, 8, e2960. [Google Scholar] [CrossRef]
- Borgese, N.; Coy-Vergara, J.; Colombo, S.F.; Schwappach, B. The Ways of Tails: The GET Pathway and more. Protein J. 2019, 38, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Bram, R.J.; Crabtree, G.R. Calcium signalling in T cells stimulated by a cyclophilin B-binding protein. Nature 1994, 371, 355–358. [Google Scholar] [CrossRef]
- Tovey, S.C.; Bootman, M.D.; Lipp, P.; Berridge, M.J.; Bram, R.J. Calcium-modulating cyclophilin ligand desensitizes hormone-evoked calcium release. Biochem. Biophys. Res. Commun. 2000, 276, 97–100. [Google Scholar] [CrossRef]
- Holloway, M.P.; Bram, R.J. Co-localization of calcium-modulating cyclophilin ligand with intracellular calcium pools. J. Biol. Chem. 1998, 273, 16346–16350. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, R.; Tajes, M.; Vazquez-Carrera, M. The NR4A subfamily of nuclear receptors: Potential new therapeutic targets for the treatment of inflammatory diseases. Expert Opin. Ther. Targets 2017, 21, 291–304. [Google Scholar] [CrossRef]
- Blackwell, K.A.; Raisz, L.G.; Pilbeam, C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010, 21, 294–301. [Google Scholar] [CrossRef] [PubMed]
- D’Eufemia, P.; Finocchiaro, R.; Celli, M.; Zambrano, A.; Tetti, M.; Villani, C.; Persiani, P.; Mari, E.; Zicari, A. High levels of serum prostaglandin E2 in children with osteogenesis imperfecta are reduced by neridronate treatment. Pediatr. Res. 2008, 63, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Velaphi, S.; Cilliers, A.; Beckh-Arnold, E.; Mokhachane, M.; Mphahlele, R.; Pettifor, J. Cortical hyperostosis in an infant on prolonged prostaglandin infusion: Case report and literature review. J. Perinatol. 2004, 24, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Estes, K.; Nowicki, M.; Bishop, P. Cortical hyperostosis secondary to prostaglandin E1 therapy. J. Pediatr. 2007, 151, 441. [Google Scholar] [CrossRef]
- Cao, Y.J.; Wei, Z.; Zhang, H.; Zhang, Z.L. Expanding the Clinical Spectrum of Osteogenesis Imperfecta Type V: 13 Additional Patients and Review. Front. Endocrinol. 2019, 10, 375. [Google Scholar] [CrossRef]
- Simon, A.M.; Manigrasso, M.B.; O’Connor, J.P. Cyclo-oxygenase 2 function is essential for bone fracture healing. J. Bone Miner. Res 2002, 17, 963–976. [Google Scholar] [CrossRef]
- Zhang, X.; Schwarz, E.M.; Young, D.A.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Investig. 2002, 109, 1405–1415. [Google Scholar] [CrossRef]
- Chen, H.; Hu, B.; Lv, X.; Zhu, S.; Zhen, G.; Wan, M.; Jain, A.; Gao, B.; Chai, Y.; Yang, M.; et al. Prostaglandin E2 mediates sensory nerve regulation of bone homeostasis. Nat. Commun. 2019, 10, 181. [Google Scholar] [CrossRef]
- Hu, B.; Lv, X.; Chen, H.; Xue, P.; Gao, B.; Wang, X.; Zhen, G.; Crane, J.L.; Pan, D.; Liu, S.; et al. Sensory nerves regulate mesenchymal stromal cell lineage commitment by tuning sympathetic tones. J. Clin. Investig. 2020, 130, 3483–3498. [Google Scholar] [CrossRef]
- Celil Aydemir, A.B.; Minematsu, H.; Gardner, T.R.; Kim, K.O.; Ahn, J.M.; Lee, F.Y. Nuclear factor of activated T cells mediates fluid shear stress- and tensile strain-induced Cox2 in human and murine bone cells. Bone 2010, 46, 167–175. [Google Scholar] [CrossRef]
- Xu, M.; Choudhary, S.; Voznesensky, O.; Gao, Q.; Adams, D.; Diaz-Doran, V.; Wu, Q.; Goltzman, D.; Raisz, L.G.; Pilbeam, C.C. Basal bone phenotype and increased anabolic responses to intermittent parathyroid hormone in healthy male COX-2 knockout mice. Bone 2010, 47, 341–352. [Google Scholar] [CrossRef]
- Blaeser, F.; Ho, N.; Prywes, R.; Chatila, T.A. Ca2+-dependent gene expression mediated by MEF2 transcription factors. J. Biol. Chem. 2000, 275, 197–209. [Google Scholar] [CrossRef]
- Li, X.; Wei, W.; Huynh, H.; Zuo, H.; Wang, X.; Wan, Y. Nur77 prevents excessive osteoclastogenesis by inducing ubiquitin ligase Cbl-b to mediate NFATc1 self-limitation. eLife 2015, 4, e07217. [Google Scholar] [CrossRef]
- Rajan, S.; Jang, Y.; Kim, C.H.; Kim, W.; Toh, H.T.; Jeon, J.; Song, B.; Serra, A.; Lescar, J.; Yoo, J.Y.; et al. PGE1 and PGA1 bind to Nurr1 and activate its transcriptional function. Nat. Chem. Biol. 2020, 16, 876–886. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maranda, V.; Gaumond, M.-H.; Moffatt, P. The Osteogenesis Imperfecta Type V Mutant BRIL/IFITM5 Promotes Transcriptional Activation of MEF2, NFATc, and NR4A in Osteoblasts. Int. J. Mol. Sci. 2022, 23, 2148. https://doi.org/10.3390/ijms23042148
Maranda V, Gaumond M-H, Moffatt P. The Osteogenesis Imperfecta Type V Mutant BRIL/IFITM5 Promotes Transcriptional Activation of MEF2, NFATc, and NR4A in Osteoblasts. International Journal of Molecular Sciences. 2022; 23(4):2148. https://doi.org/10.3390/ijms23042148
Chicago/Turabian StyleMaranda, Vincent, Marie-Hélène Gaumond, and Pierre Moffatt. 2022. "The Osteogenesis Imperfecta Type V Mutant BRIL/IFITM5 Promotes Transcriptional Activation of MEF2, NFATc, and NR4A in Osteoblasts" International Journal of Molecular Sciences 23, no. 4: 2148. https://doi.org/10.3390/ijms23042148
APA StyleMaranda, V., Gaumond, M. -H., & Moffatt, P. (2022). The Osteogenesis Imperfecta Type V Mutant BRIL/IFITM5 Promotes Transcriptional Activation of MEF2, NFATc, and NR4A in Osteoblasts. International Journal of Molecular Sciences, 23(4), 2148. https://doi.org/10.3390/ijms23042148