1. Introduction
Prostate cancer is one of the most common types of cancer in men, and the risk of developing it increases with advanced age. More than 75% of patients with prostate cancer are men over the age of 65 [
1]. According to Siegel’s statistic (2022), it is estimated that about 268,490 new cases of prostate cancer will be reported in the United States in 2022 [
2]. In most deaths associated with prostate cancer, there is a predominant type known as castration-resistant prostate cancer (CRPC) [
3]. Typical CRPC cellular mechanisms include androgen receptor (AR) overexpression, intratumoral synthesis of androgens, and the expression of shortened AR variants. [
4]. These AR variants arise from alternative splicing of cryptic exons, and their activity is mostly ligand-independent [
5]. The most abundant variant is AR-v7, which was indicated as a possible biological marker of prostate cancer development [
6] since its levels have been reported to be significantly lower in normal prostate tissue than in prostate cancer tissue, or CRPC [
5]. Prostate cancer with this splicing variant shows only a minor therapeutic response to commonly used anti-androgenic drugs (e.g., enzalutamide; ENZ) [
7].
AR belongs to the nuclear receptor superfamily. Upon androgen (ligand) binding, AR translocates to the nucleus and binds to specific responsive elements in DNA. The most vital androgens are testosterone (T), dihydrotestosterone (DHT), and dehydroepiandrosterone (DHEA) [
8]. Testosterone is the main male sex hormone and is converted to DHT by 5α-reductase enzyme. This process occurs in several target tissues, e.g., in the prostate [
8,
9]. Because both hormones bind to the AR, they are the main AR ligands, with DHT being more potent to AR than T [
9]. AR activation through DHT is also essential for normal prostate development and function [
10]. Imbalanced androgen secretion is linked with the occurrence of several associated diseases or syndromes, e.g., congenital lipoid adrenal hyperplasia, pseudohermaphroditism, or even affecting the development of prostate cancer [
8,
10,
11]. Androgens modulate a wide range of biological responses in the human body through AR and AR became a significant therapeutic target in prostate cancer treatment [
8]. Nowadays, the most common group of therapeutic drugs (i.e., direct approach) used in prostate cancer treatment are AR antagonists (e.g., bicalutamide, nilutamide, or ENZ) [
12].
More than a decade ago, a different prostate cancer therapeutic strategy was suggested, assuming suppression of AR function through activation of the aryl hydrocarbon receptor (AhR), i.e., an indirect approach. Similarl to AR, upon ligand binding, AhR translocates into the nucleus, where it binds to a specific xenobiotic-responsive element in DNA, with a consequent increase in the expression of the target genes [
13,
14]. AhR has been primarily associated with biotransformation, but its ability to affect the function of other receptor pathways has also been described [
15,
16]. This receptor binds a wide number of endo- or exogenous molecules, with polyaromatic hydrocarbons (PAHs) or dioxins, such as, 2,3,7,8-tetrachlondibenzo-p-dioxin (TCDD), being the most potent exogenous ligands. Some PAHs have also been studied for anti-estrogenic and anti-androgenic effects [
17].
An in vivo connection between AhR and AR in relation to prostate cancer was suggested in the study by Fritz et al. 2007, who used three different genotypes of mice, namely
AhR+/+,
AhR+/−, and
AhR−/−, and studied the impact on prostate cancer development [
18]. The obtained results suggested that the presence of AhR inhibits prostate carcinogenesis, and the model of prostate cancer development based on the genotype of the mice was as follows:
AhR+/+ < AhR+/− <
AhR−/−, where
AhR+/+ mice had the lowest incidence of prostate cancer development compared to
AhR−/− mice (16% vs. 60%) [
18].
Interestingly, only a limited number of studies have dealt with possible crosstalk between AhR and AR in human prostate cells. One such cross-talk can represent AhR-mediated AR degradation, since AhR has been demonstrated as a ligand-dependent E3 ubiquitin ligase that induces proteasomal degradation of AR in the androgen-sensitive prostate cancer cell line LNCaP [
19]. However, it appears that the effect differs between cell lines since the castration-resistant type (C4-2 cell line), TCDD did not induce AR degradation through AhR activation [
20]. The AhR and AR protein levels were significantly decreased after the treatment of LNCaP cells with another potent AhR agonist from the PAH group, 3-methylcholanthrene (3MC) [
21]. The anti-androgenic effects of AhR ligands were also described for various AhR agonists, such as chrysene, benzo[k]fluoranthene, and benzoapyrene (B[a]P) but not for anthracene or pyrene. Moreover, listed AhR agonists with anti-androgenic effects also increased c-fos and c-jun mRNA levels [
17].
The study of Arabnezhad et al. 2020 considered the effects of endogenously activated AhR on AR [
22]. The prostate cell line LNCaP was treated with 6-formylindolo [3,2-b]carbazole (FICZ) in the presence or absence of the AR ligand testosterone. After treatment, mRNA (
AR,
KLK2,
TMPRSS2, and
PSA), PSA protein levels, and DHT levels were evaluated as the end points. The study confirmed that FICZ induced
CYP1A1 activity, which is a marker of AhR activation. In addition, a significant decrease in AR-target gene mRNA expression was observed with the combination of FICZ (50 nM) and testosterone (100 nM). PSA protein and DHT levels were reduced after treatment with FICZ + T. However, no decrease was observed in the absence of testosterone. The obtained results indicated that AhR plays an important role in AR signaling and that FICZ has anti-androgenic effects through the AhR/AR pathway [
22]. Moreover, Morrow et al. 2004 study suggested that the stability of AR protein is strictly dependent on AhR ligand and that AhR/AR inhibitory crosstalk is more likely promoter specific [
23]. Using a co-immunoprecipitation assay, it was demonstrated that AhR forms a complex with AR and that this process is fundamentally facilitated by DHT. Furthermore, the presence of DHT decreased 3MC-induced transcription of
CYP1A1 [
21].
Interestingly, the concept of AhR-mediated degradation of AR was described for other compounds, namely Icaritin [
24] and Carbidopa [
25]. Icaritin is one of the major metabolites of the compound Icariin [
26,
27]. Both of these substances are naturally occurring polyphenols, which can be found in plants of the genus
Epimedium [
27]. Carbidopa is a decarboxylase inhibitor used to treat Parkinson’s disease [
28]. Both of these compounds have been shown to induce AhR activation with consequent degradation of AR [
24,
25]. However, while Icaritin induced this effect in LNCaP, C4-2 and 22Rv1 cells, Carbidopa was demonstrated to have such an effect in LNCaP cells only. Moreover, these compounds suppressed the tumor proliferation of LNCaP implanted cells in nude mice [
24,
25].
Therefore, available studies strongly indicate the presence of AhR/AR crosstalk in prostate cancer cells, as various AhR ligands were reported to inhibit prostate cancer development, probably through several different mechanisms [
22]. Published data suggest that only strong and potent AhR ligands are capable of inducing AR degradation through AhR activation [
19].
Recently, our research team observed that the group of mostly synthetic compounds with an indole scaffold has the ability to activate AhR in hepatocarcinoma cells (AZ-AhR, derived from the parental HepG2 cell line) [
29] with efficacy comparable to TCDD (5 nM).
Therefore, the objective of this study was to determine whether these indoles have the ability to suppress AR function through AhR activation and thus slow the proliferation of cancer cells.
3. Discussion
The main goal of this study was to demonstrate if AhR activation by certain indoles can result in the degradation or at least suppression of AR activity in prostate cancer cells. This assumption was based on the observations that strong AhR ligands degraded AR in the LNCaP prostate cell line [
15,
19] and that two other compounds, namely Icaritin [
24] and Carbidopa [
25], which could activate AhR, suppressed prostate cancer via AhR-mediated degradation of AR. Effects of these compounds and indoles from our study on AhR and AR pathways are summarized in
Table 2. The tested indoles were selected on the basis of a recent report by our research group, in which methyl- and methoxyindoles were identified as AhR agonists [
29]. In the human hepatocarcinoma AZ-AhR cell line, all indoles activated AhR to some extent, and the strongest ones were reported to be 4 MI, 5 MI, 6 MI, 2,5 DMI, and 7MeO [
29]. Similarly, all tested indoles activated AhR in the 22Rv1 prostate cell line after 24 h with efficacy comparable to TCDD, which drastically less activated AhR (5-fold, 10 nM) than AZ-AhR (1000-fold, 5 nM). A partial explanation of such a huge difference lies in the significantly lower amount of AhR protein in the prostate (22Rv1) than in AZ-AhR cells (personal observation).
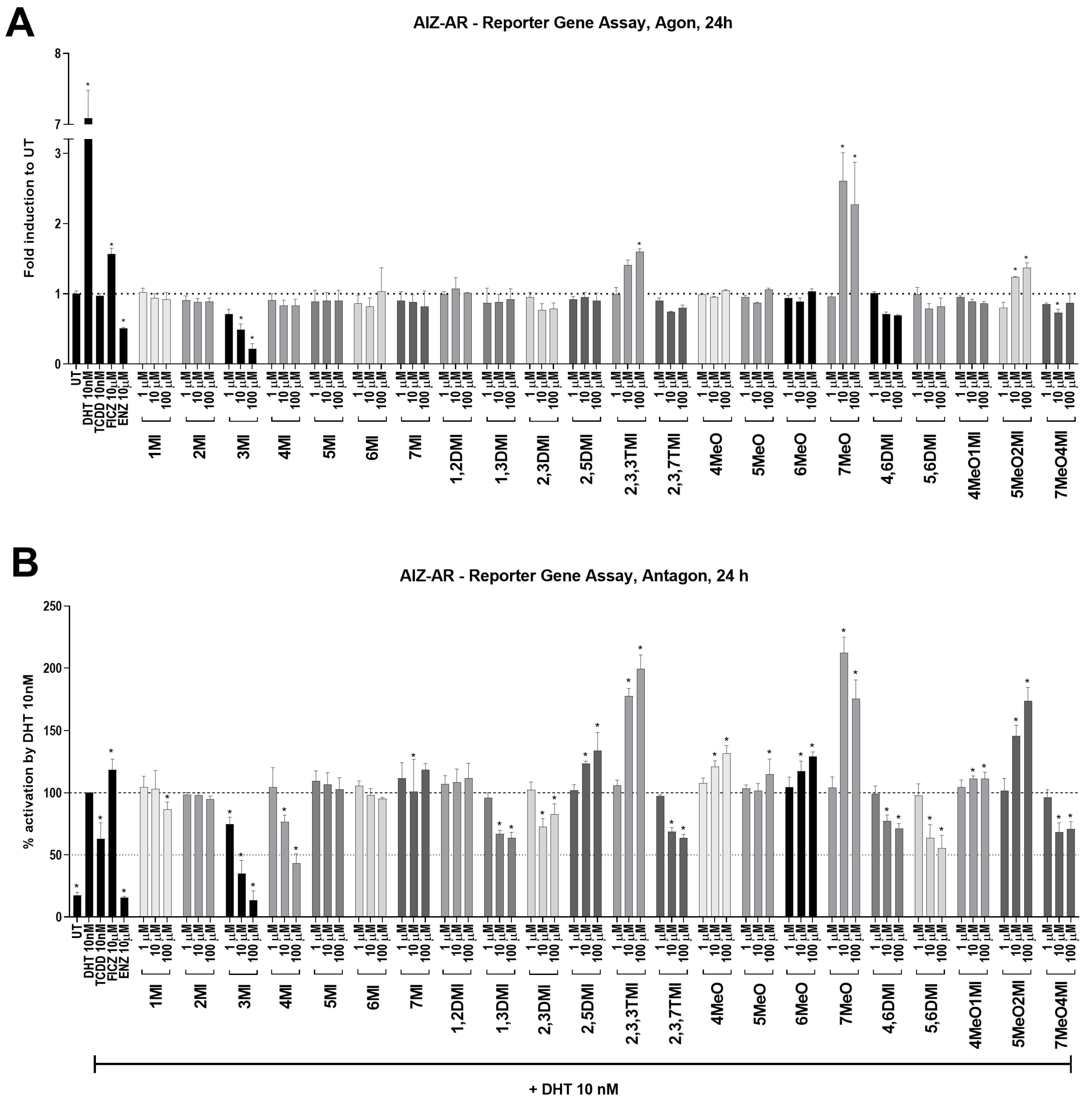
Despite the fact that all indoles activated AhR comparably to TCDD (
Figure 1), only some of them were able to suppress AR activity (
Figure 2) and consequently AR-target gene expression (
Figure 3). Interestingly, typical high affinity AhR ligands, TCDD and FICZ, had a opposite impact on the DHT-inducible AR-mediated luciferase activity (
Figure 2), despite concentration-dependent induction of AhR-mediated luciferase activity (
Supplementary S1). Furthermore, selected indoles that suppressed AR activity inhibited DHT-inducible AR binding to the
KLK3 promoter to some extent, suggesting an impact on the initial transcription event. Additionally, these indoles suppressed AR mRNA levels, which were consequently reflected in AR protein levels. This suggests a different type of mechanism from that initially expected. Additionally, despite very strong AhR activation next to TCDD, most of the indoles have not been demonstrated to be true AhR ligands so far, and thus, the lack of AR degradation effect may stand beyond this fact. This is consistent with the original observation by Ohtake et al., 2007 [
19], who found that only full agonists such as TCDD, BaP, and 3MC could degrade AR but weaker ligands (e.g., indirubin–indole scaffold, β-naphthoflavone) could not.
Further, next to the nature of a ligand/agonist of AhR, probably a specific environment plays a role since the study by Ohtake et al., 2007 [
19] used LNCaP cells treated for 3 h in medium with 0.2% charcoal-stripped serum, while we used 22Rv1 cells in medium with 10% regular serum and added 10 nM DHT to mimic basal androgen secretion in the human body. Another difference between these two cell lines lies in the presence of AR variants, where LNCaP has one (AR-fl), while 22Rv1 has two (AR-fl, AR-v7) [
5]. Furthermore, AR-v7 displays nuclear localization even in the absence of androgen and acts as a transcription factor [
5].
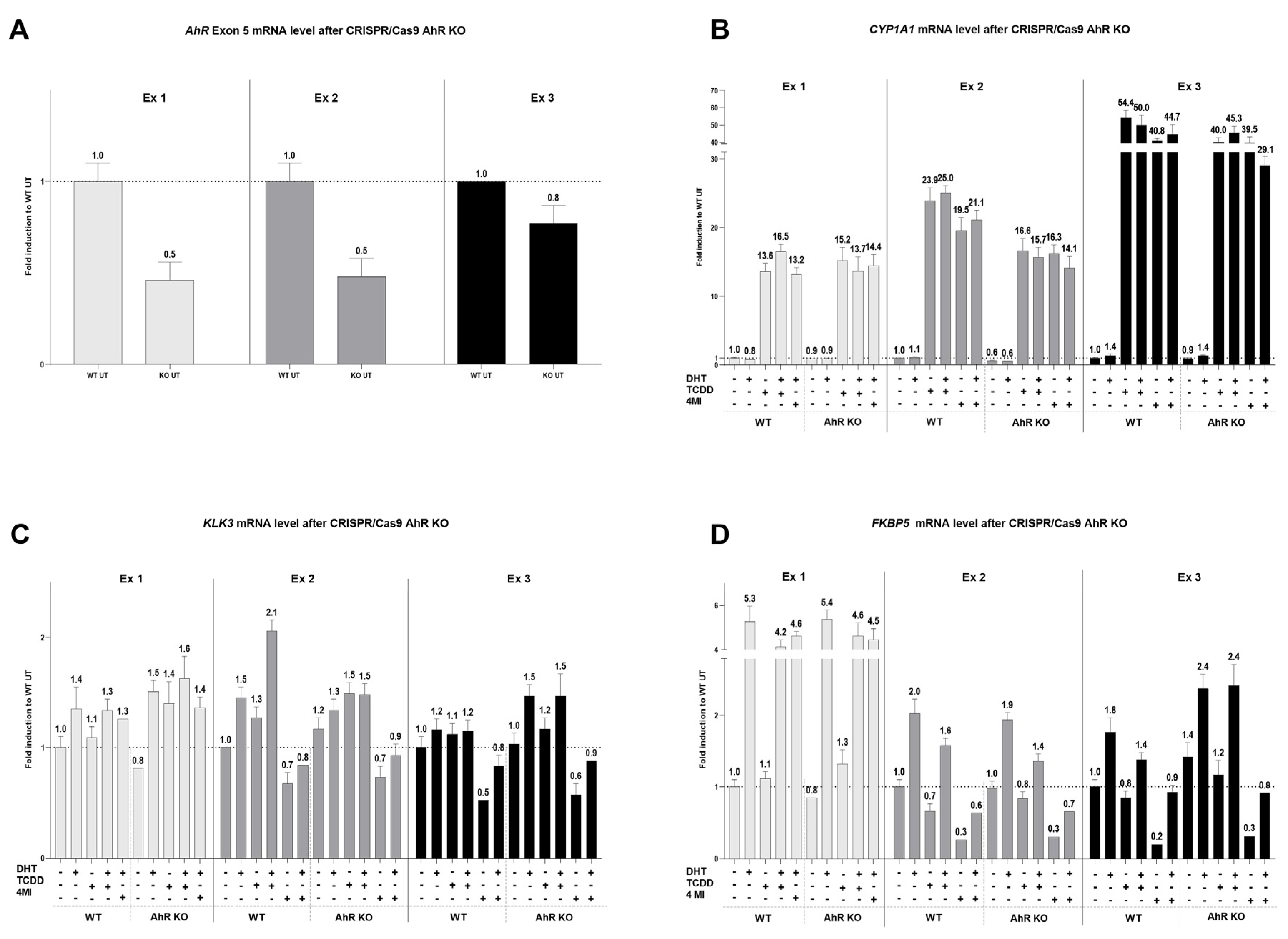
Additionally, it seems that different types of prostate cancer cells have shown different sensitivity to affecting the AR pathways through AhR activation. The AR degradation process through AhR activation was not identified in all types of prostate cancer cells. In the androgen-sensitive LNCaP cell line, which was isolated from the left supraclavicular lymph [
32], the mechanism of AR degradation through AhR activation by a strong ligand has been observed multiple times [
20,
22]. LNCaP cells are classified as AR and AhR-positive [
21]. However, the same AhR ligand (TCDD) did not induce AR degradation in the castration-resistant C4-2 cell line [
20], which was derived from the parental LNCaP [
33]. This may be due to the fact that different types of prostate cancer are diverse in AhR and AR receptor content, and subcellular localization, and that AhR presence may affect AR phosphorylation status [
34]. This is probably also reflected in our data, since ChIP results demonstrated a decrease in AR binding to the
KLK3 promoter after 90 min (
Figure 4), while AR protein levels (
Figure 5) were almost unaffected by most of the tested indoles after 24 h with the 10 µM concentrations used in ChIP. Thus, our results suggest a mix of potential post-translational modification of AR or affected interaction with other transcription partners. This consequently leads to a decrease in AR binding to the
KLK3 promoter and a decrease in the mRNA levels of AR-target genes.
Probably the most interesting finding is the impact of 3MI (skatole) on AR activity and cell viability. Skatole is naturally a product of the intestinal microbiota and can be found in wide range of concentrations in the serum at least of patients with hepatic encephalopathy [
35]. Unfortunately, it is not known what the average concentration of skatole in the serum of healthy individuals is. Therefore, our results may suggest a hypothesis that men with detectable skatole concentration in the serum are less likely to develop/have AR-dependent prostate cancer. The proposition of this hypothesis is based on the observation of a decline in all AR-mediated parameters we studied, as well as on the strongest decline in the Crystal violet assay, which is a reflection of the proliferative capacity of the cells.
Furthermore, we observed a visible morphological change in 22Rv1 cells at the end of skatole treatment (personal observation). Thus, it is likely that skatole triggers some sort of apoptotic event and apparently has more molecular targets inside the cells. Our assumption is based on the observation of different skatole action in relation to other indoles as it reduced rather than induced
AhRR mRNA (
Figure 3B) and consistently induced concentration-dependently
AhR mRNA (personal observation). The expression of
AhRR and
AhR is regulated by NF-kB [
36], and the combination of genotoxic action, demonstrated for skatole in bronchial cells [
37] (that is, activation of p53), and activation of NF-kB could explain these differences. However, this must be revealed in future research as a possible relationship (if any) between skatole blood level and prostate cancer incidence in men.
We believe that skatole deserves further investigation in the future in relation to prostate cancer.
The hypothesis of AR degradation through AhR activation by selected indolic compounds was tested. All 22 tested indoles displayed promising AhR-activating potential in the newly developed 22AhRv1 reporter cell line. However, only 8 of them were able to supress DHT-induced AR activation, namely 3MI, 4MI, 1,3DMI, 2,3DMI, 2,3,7TMI, 4,6DMI, 5,6DMI, and 7MeO4MI. At the mRNA level, 8 selected indoles increased CYP1A1 expression and significantly reduced DHT-inducible KLK3 and FKBP5 expression. Moreover, following ChIP analysis revealed reduced DHT-inducible binding of AR to the KLK3 promoter by selected indoles. The effect of the strongest AhR activator in this study, 4MI, was further monitored using transient transfection with the CRISPR/Cas9 AhR KO plasmid. However, after AhR KO, no change in AR-targets was observed. To conclude, some tested indoles showed the ability to suppress DHT-induced AR activation; however, this phenomenon does not seem to be associated with AhR activation. Therefore, the hypothesis of AR degradation via AhR activation for selected indoles has not been confirmed.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}