
Increased Cell Proliferation as a Key Event in Chemical Carcinogenesis: Application in an Integrated Approach for the Testing and Assessment of Non-Genotoxic Carcinogenesis

 ,
,
Abstract
:1. Introduction
2. Assessing Cell Proliferation In Vivo and In Vitro
2.1. General Aspects
2.1.1. Nucleoside Analog Incorporation into Newly Synthesized DNA
2.1.2. Cell Cycle-Associated Protein Expression and Corresponding mRNA
2.1.3. The Tools of Choice
2.1.4. Target Organs and Target Cell Types: Some Case Examples
2.2. Challenges of the In Vivo Evaluation of Cell Proliferation
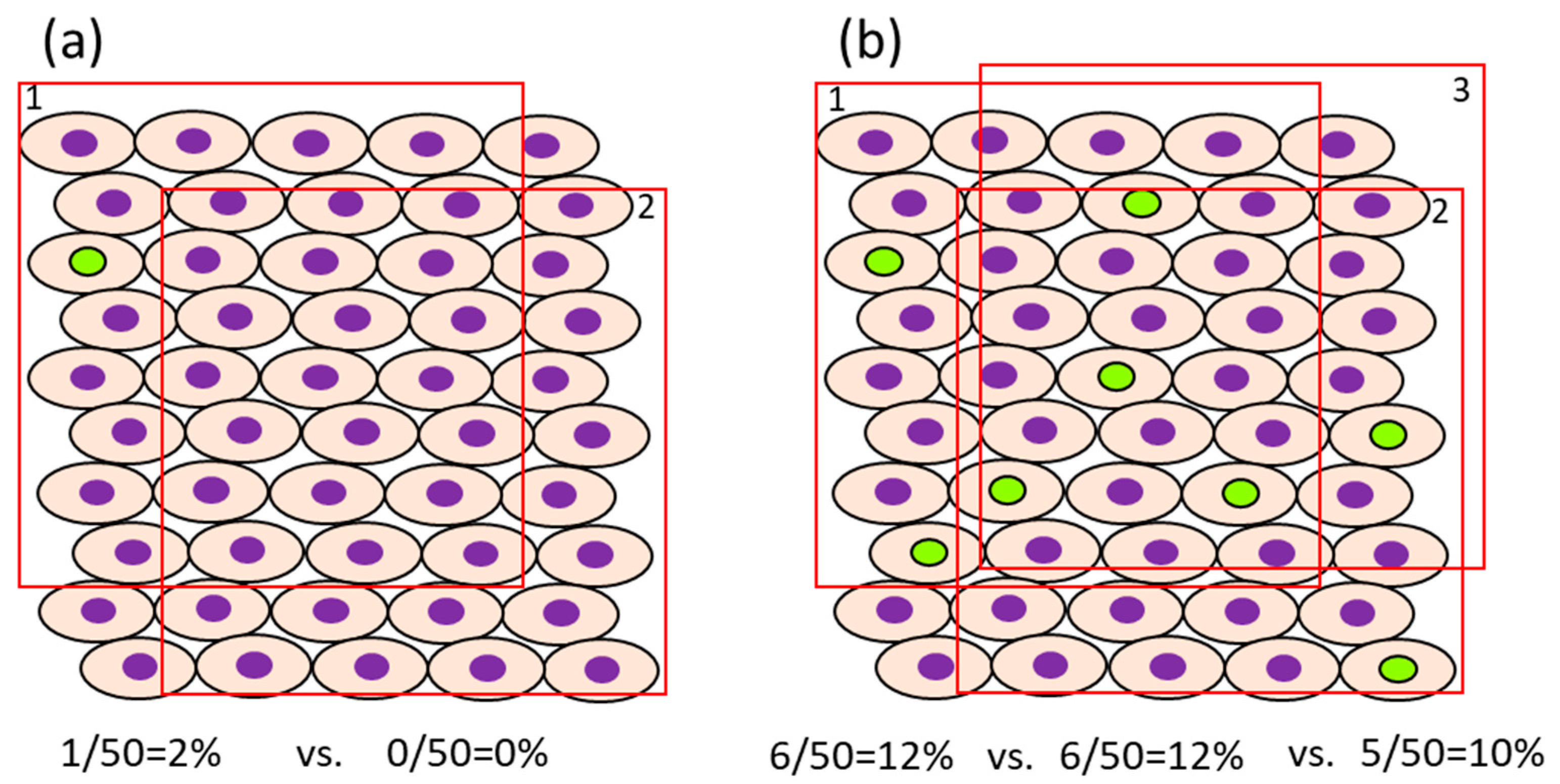
- In case the proliferation rate of the target cells is fast, it may be possible to reduce the number of cells that need to be counted. For example, in the normal colon of rats injected i.p. with BrdU 1 or 2 h before sacrifice, approximately 4.1–6.7/pit [54] or 12.8–15.8% [55] were positive, respectively, when they were counted in 12 or 10 crypts. The number of cells, as a denominator of the evaluation, differs between organs/cells with rapid cell proliferation (e.g., intestine) and those with slow cell proliferation (e.g., liver and bladder) [32]. For example, the ratio of BrdU-positive cells in the normal liver and bladder of rats injected i.p. with BrdU 1 h before sacrifice is about 0.28–0.48% [54] and 0.06–0.32% [54,56], respectively. In such cases, for sufficient statistical power, at least 1000–3000 cells should be evaluated for comparison. The impact of the error in the number of positive cells depending on the selection of view may become small if the number of positive cells is high. However, when the ratio of positive cells is low, fewer counts will result in a greater error due to the difference depending on the field of view (Figure 2). Also, to avoid selection bias, it is recommended that the slides be coded so that the individual counting the cells is blinded to the group the sample is from. The evaluation fields should be as similar as possible with respect to histological location.
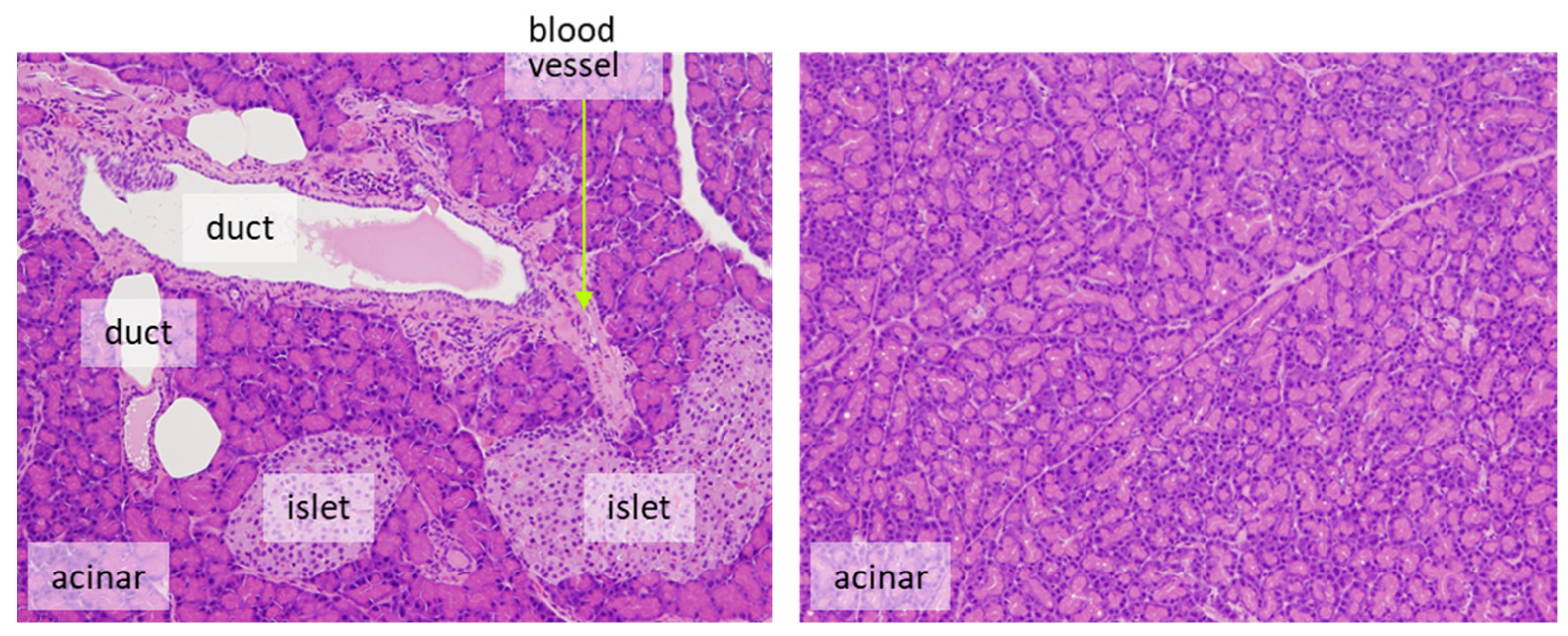
- It is desirable to count only target cells and not multiple cell types. For example, for the evaluation of proliferation in pancreatic endocrine cells, only islet cells should be assessed. Exocrine tumors can arise from ductular or acinar cells. Although the phenomenon is rarely seen in rats, the human pancreas is more likely to develop carcinogenesis from duct-derived cells than from islets or acinar cells. If there are reasonable animal models for human pancreatic cancer, it is appropriate to evaluate only the ductal cells (Figure 3). However, this may be decided on a case-by-case basis.
- 3.
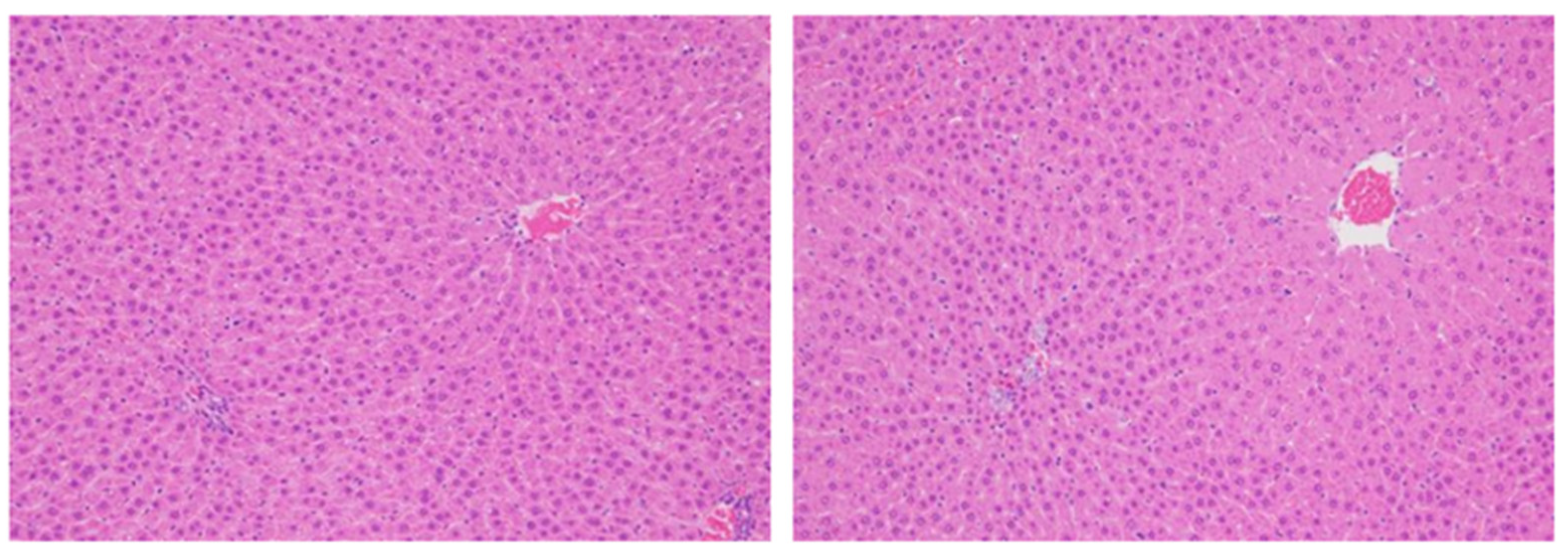
- In principle, it is considered more accurate to evaluate proliferation using the number of cells as the denominator, but when cells exist at the same density, it is also possible to examine per unit area. Consideration is nevertheless required when the size or the density of target cells differs amongst groups. For example, if there is hepatocyte hypertrophy, the number of cells per unit area will become smaller, the denominator will be overestimated, and the positive ratio will become smaller than it is (Figure 4).
- 4.
- If there is a difference in cell proliferation across the organ, it would be necessary to evaluate the fields evenly to reflect the whole organ. For example, the epithelial cells in the gastrointestinal tract and urinary bladder have certain proliferative zones consisting of immature cells located mostly in the basal layer of the urothelium or in the crypts of the intestine, but middle height in the mucosal glands in the stomach. A bias may occur in the ratio between the different layers depending on how the organ is cut, so care must be taken to make the sectioning representative of the normal distribution of target cells (Figure 5). To avoid this bias, a greater number of cells should be evaluated compared to more homogenous tissues.
- 5.
- When tissues with a mixture of lesions are anticipated, it is important to evaluate cell proliferation in each of the histological lesions such as normal-like areas, hyperplasia, benign tumors, and malignant tumors. If the histopathological lesion is not considered, the evaluation field should reflect the existing lesion as a whole and care must be taken to avoid bias. Hyperplastic areas indicate higher cell proliferation than normal-like areas, usually with an increased rate of proliferation as well as an increased number of cells.
- 6.
- Evaluation of BrdU (or/and 3H-labeling) indices should be compared to a control group administered under the same conditions. Factors such as the time after administration and the time of day (diurnal changes), and exposure concentration may impact results. Basically, if enough BrdU or 3H is administered, even if the amount of uptake into each nucleus during the DNA synthesis varies, the mitotic phase changes. The labelling reagents may not be taken up by cells outside the S phase, resulting in inconsistent labeling values. Ki-67 evaluation may also be affected by the duration of the fixation time and the specimen preparation conditions, for example, so the comparison to the concurrent control group is crucial. Samples for the determination of Ki-67 should ideally not be left in the fixative for more than 72 h before trimming and embedding.
- 7.
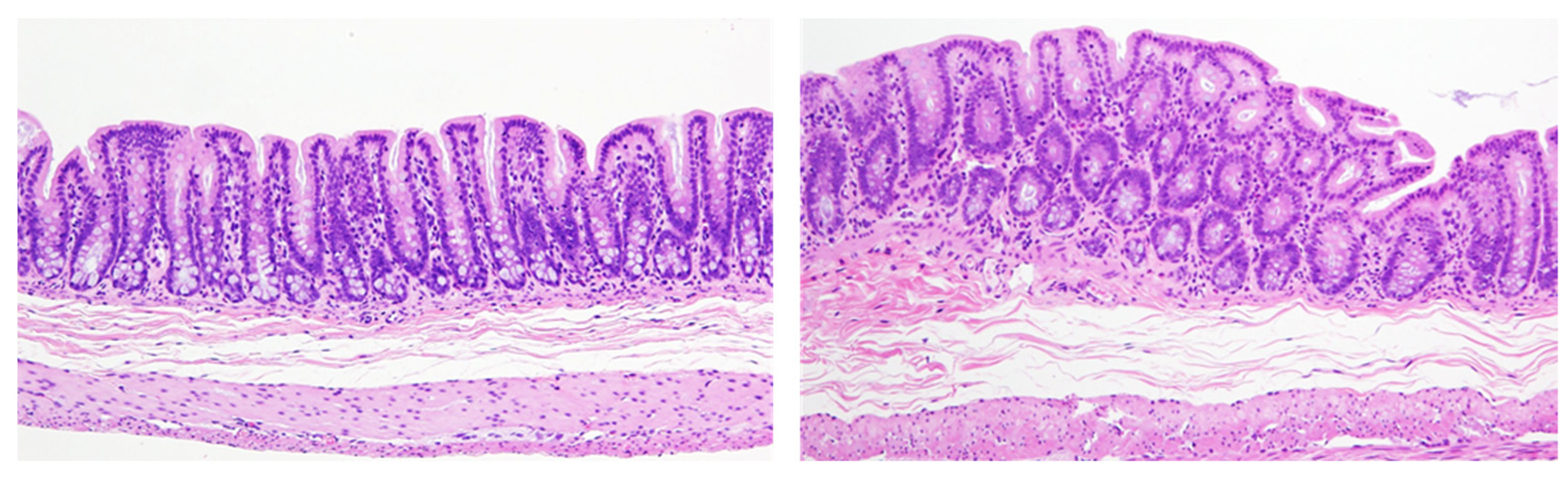
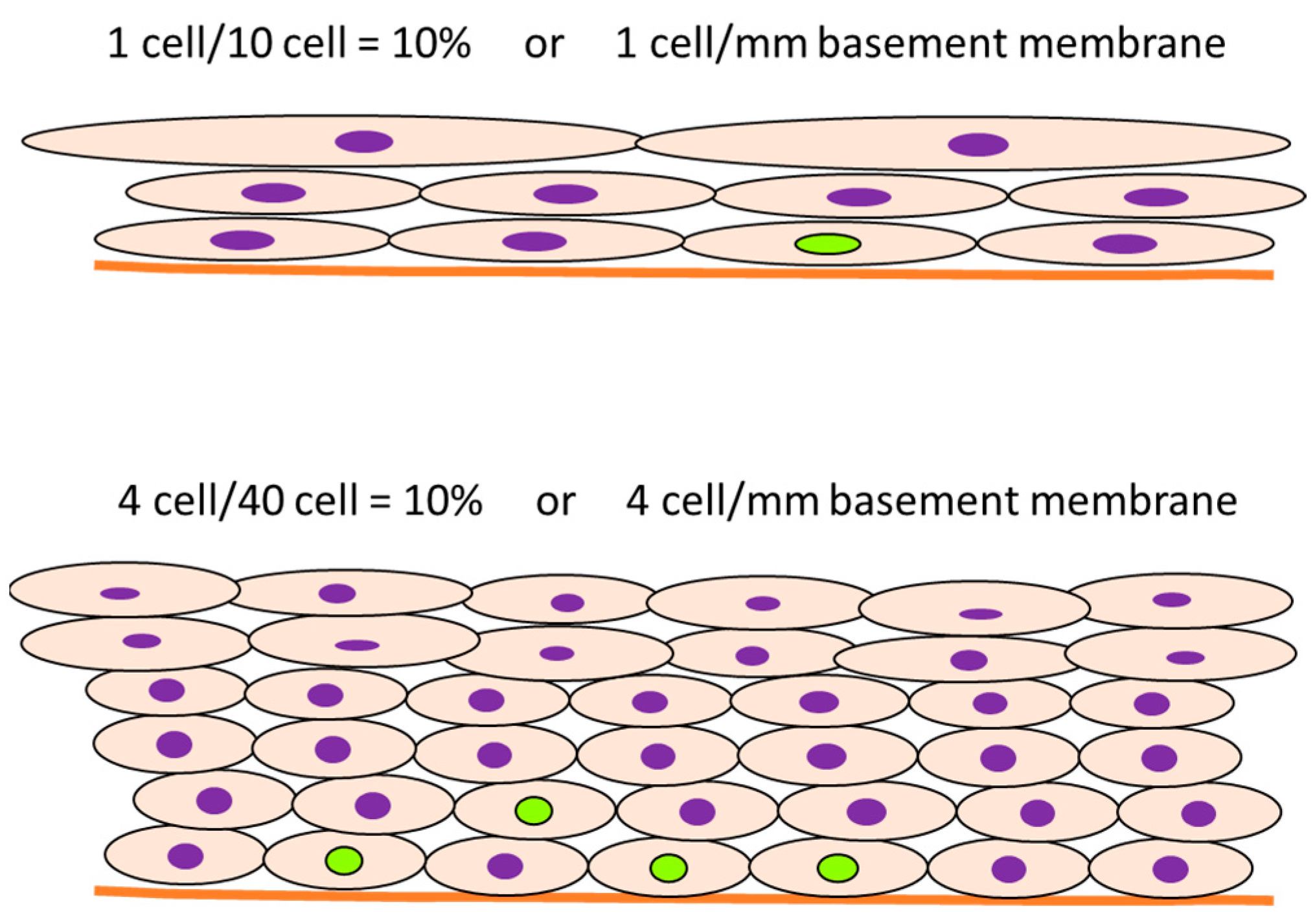
- In some cases, the cell proliferation rate is transiently induced in the initial period of administration of the test chemical, and the labeling rate increases, after which the proliferation ratio returns to normal levels. However, an increase in cell number results in an increase in the denominator, and the number of proliferating cells is still high even though the labeling rate looks decreased. For example, the meaning may be different between 10% positive normal-looking urothelial cells in three layers and 10% positive urothelial cells in six layers due to simple hyperplasia with increased cell density. In that case, the number of positive cells per unit basement membrane length is more than doubled and may be of different significance (Figure 6).
2.3. Challenges and Opportunities for the In Vitro Evaluation of Cell Proliferation
2.4. “Readiness” and Appropriateness Evaluation of Cell Proliferation Assays
3. The Value of Adding “-Omics” to In Vitro/In Vivo Cell Proliferation Assays
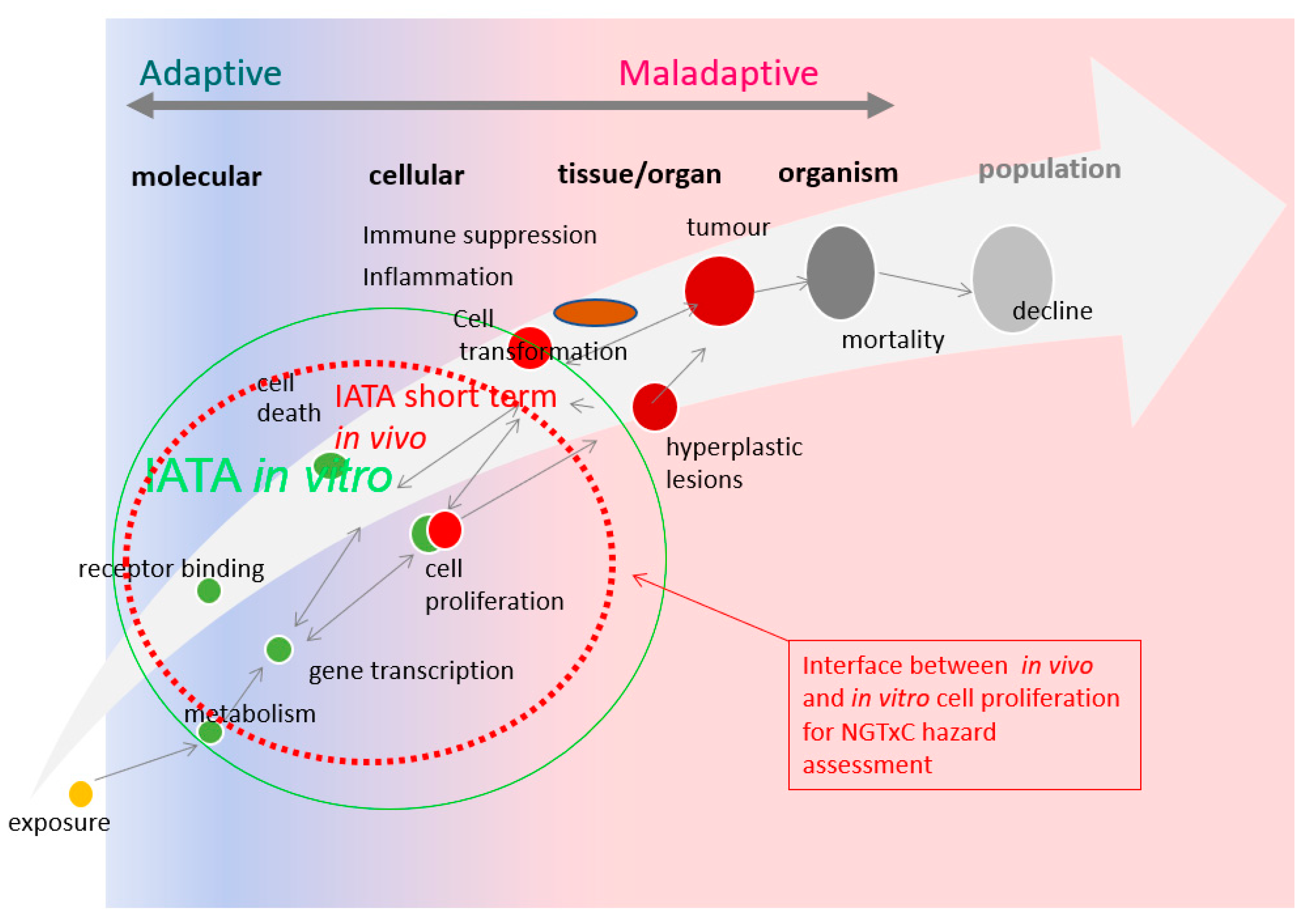
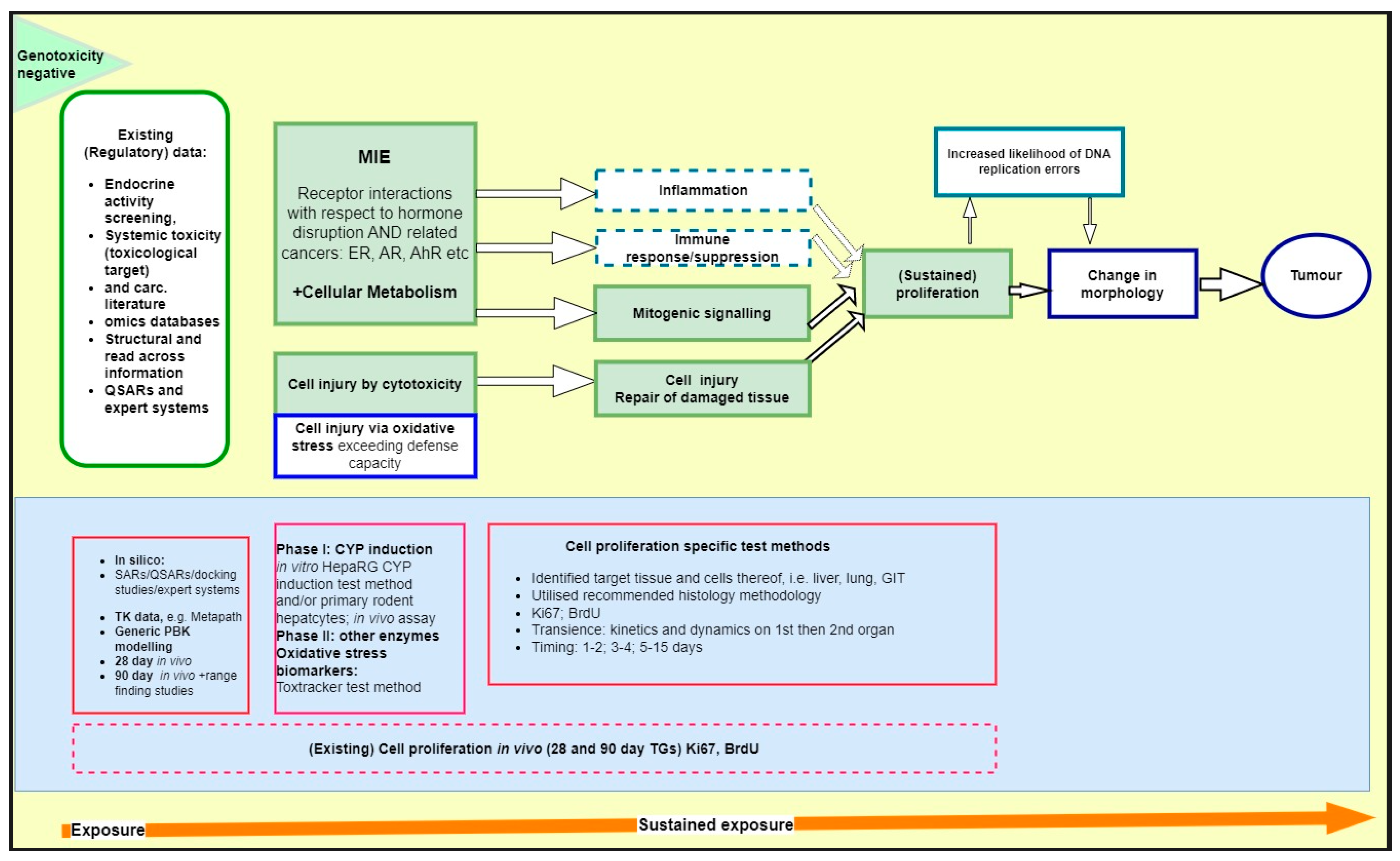
4. Application of Cell Proliferation Assay Tools in the NGTxC IATA: Proposed Way Forward
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADME | Absorption–distribution–metabolism–excretion |
| AhR | Aryl hydrocarbon receptor |
| AIME | Alginate immobilization of metabolic enzymes |
| AR | Androgen receptor |
| ATP | Adenosine triphosphate |
| BrdU | Tritiated thymidine, 5-bromo-2’-deoxyuridine |
| BW | Body weight |
| CAR | Constitutive androstane receptor |
| CCP | Cell cycle proliferation |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CYP | Cytochrome P450 |
| 14C-Thy | 14Carbon-labelled thymidine |
| ECHA | European Chemicals Agency |
| EFSA | European Food Safety Authority |
| EGF | Epidermal growth factor |
| ER | Estrogen receptor |
| FANFT | N-(4-(5-Nitro-2-furyl)-2-thiazolyl)formamide |
| FFPE | Formalin-fixed paraffin-embedded |
| GARDTM | Genomic Allergen Rapid Detection (in vitro platform for safety assessment of chemicals) |
| GIT | Gastrointestinal tract |
| H2O2 | Hydrogen peroxide |
| 3H-Thy | Tritiated thymidine |
| HTTr | High-throughput transcriptomics |
| IATA | Integrated approaches to testing and assessment |
| ICH | International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use |
| i.p. | Intraperitoneal |
| IPCS | International Program on Chemical Safety |
| i.v. | Intravenous |
| Ki-67 | Nuclear antigen expressed in all phases of the cell cycle and mitosis |
| LW | Liver weight |
| MIE | Molecular initiating event |
| MoA | Mode of action |
| MSigDB | Molecular Signatures Database |
| NAMs | New approach methods |
| NGTxC | Non-genotoxic carcinogens |
| OECD | Organization for Economic Cooperation and Development |
| PBK | Physiologically based kinetic |
| PCNA | Proliferating cell nuclear antigen |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PXR | Pregnane X receptor |
| QSAR | Quantitative structure activity relationship |
| ReCAAP | Rethinking Carcinogenicity Assessment for Agrochemicals Project |
| s.c. | Subcutaneous |
| TCGA | The Cancer Genome Atlas |
| TG | Test guideline |
| TGx-DDI | Toxicogenomic DNA damage-inducing |
| T-hormone | Thyroid hormone |
| TSH | Thyroid-stimulating hormone |
| UDP-GT | Uridine 5’-diphospho-glucuronosyl transferase |
| US FDA | United States Food and Drug Administration |
| WHO | World Health Organization |
| Wyeth (WY) | 4-chloro-6-(2,3-xylidino)-pyrimidilnylthioacetic acid |
References
- Armitage, P.; Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 1954, 8, 1–12. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Moolgavkar, S.H.; Knudson, A.G., Jr. Mutation and cancer: A model for human carcinogenesis. J. Natl. Cancer Inst. 1981, 66, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, R.E.; Ellwein, L.B.; Cohen, S.M. A general probabilistic model of carcinogenesis: Analysis of experimental urinary bladder cancer. Carcinogenesis 1984, 5, 437–445. [Google Scholar] [CrossRef]
- Cohen, S.M.; Ellwein, L.B. Cell proliferation in carcinogenesis. Science 1990, 249, 1007–1011. [Google Scholar] [CrossRef]
- Cohen, S.M.; Ellwein, L.B. Genetic errors, cell proliferation, and carcinogenesis. Cancer Res. 1991, 51, 6493–6505. [Google Scholar]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Cohen, S.M.; Boobis, A.R.; Dellarco, V.L.; Doe, J.E.; Fenner-Crisp, P.A.; Moretto, A.; Pastoor, T.P.; Schoeny, R.S.; Seed, J.G.; Wolf, D.C. Chemical carcinogenicity revisited 3: Risk assessment of carcinogenic potential based on the current state of knowledge of carcinogenesis in humans. Regul. Toxicol. Pharmacol. 2019, 103, 100–105. [Google Scholar] [CrossRef]
- Wolf, D.C.; Cohen, S.M.; Boobis, A.R.; Dellarco, V.L.; Fenner-Crisp, P.A.; Moretto, A.; Pastoor, T.P.; Schoeny, R.S.; Seed, J.G.; Doe, J.E. Chemical carcinogenicity revisited 1: A unified theory of carcinogenicity based on contemporary knowledge. Regul. Toxicol. Pharmacol. 2019, 103, 86–92. [Google Scholar] [CrossRef]
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 2011, 120 (Suppl. S1), S130–S145. [Google Scholar] [CrossRef] [PubMed]
- Dragan, Y.P.; Bidlack, W.R.; Cohen, S.M.; Goldsworthy, T.L.; Hard, G.C.; Howard, P.C.; Riley, R.T.; Voss, K.A. Implications of apoptosis for toxicity, carcinogenicity, and risk assessment: Fumonisin B(1) as an example. Toxicol. Sci. 2001, 61, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Meek, M.E.; Bucher, J.R.; Cohen, S.M.; Dellarco, V.; Hill, R.N.; Lehman-McKeeman, L.D.; Longfellow, D.G.; Pastoor, T.; Seed, J.; Patton, D.E. A framework for human relevance analysis of information on carcinogenic modes of action. Crit. Rev. Toxicol. 2003, 33, 591–653. [Google Scholar] [CrossRef]
- Elcombe, C.R.; Peffer, R.C.; Wolf, D.C.; Bailey, J.; Bars, R.; Bell, D.; Cattley, R.C.; Ferguson, S.S.; Geter, D.; Goetz, A.; et al. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit. Rev. Toxicol. 2014, 44, 64–82. [Google Scholar] [CrossRef]
- Yamada, T.; Cohen, S.M.; Lake, B.G. Critical evaluation of the human relevance of the mode of action for rodent liver tumor formation by activators of the constitutive androstane receptor (CAR). Crit. Rev. Toxicol. 2021, 51, 373–394. [Google Scholar] [CrossRef]
- Corton, J.C.; Cunningham, M.L.; Hummer, B.T.; Lau, C.; Meek, B.; Peters, J.M.; Popp, J.A.; Rhomberg, L.; Seed, J.; Klaunig, J.E. Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARalpha) as a case study. Crit. Rev. Toxicol. 2014, 44, 1–49. [Google Scholar] [CrossRef]
- Corton, J.C.; Peters, J.M.; Klaunig, J.E. The PPARalpha-dependent rodent liver tumor response is not relevant to humans: Addressing misconceptions. Arch. Toxicol. 2018, 92, 83–119. [Google Scholar] [CrossRef]
- Veltman, C.H.J.; Pennings, J.L.A.; van de Water, B.; Luijten, M. An Adverse outcome pathway network for chemically induced oxidative stress leading to (non)genotoxic carcinogenesis. Chem. Res. Toxicol. 2023, 36, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, H.; Rasinger, J.D.; Hammer, H.; Naboulsi, W.; Zabinsky, E.; Planatscher, H.; Schwarz, M.; Poetz, O.; Braeuning, A. Proteomic analysis of hepatic effects of phenobarbital in mice with humanized liver. Arch. Toxicol. 2022, 96, 2739–2754. [Google Scholar] [CrossRef] [PubMed]
- Sonich-Mullin, C.; Fielder, R.; Wiltse, J.; Baetcke, K.; Dempsey, J.; Fenner-Crisp, P.; Grant, D.; Hartley, M.; Knaap, A.; Kroese, D.; et al. IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul. Toxicol. Pharmacol. 2001, 34, 146–152. [Google Scholar] [CrossRef]
- Seed, J.; Carney, E.W.; Corley, R.A.; Crofton, K.M.; DeSesso, J.M.; Foster, P.M.; Kavlock, R.; Kimmel, G.; Klaunig, J.; Meek, M.E.; et al. Overview: Using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit. Rev. Toxicol. 2005, 35, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Boobis, A.R.; Cohen, S.M.; Dellarco, V.; McGregor, D.; Meek, M.E.; Vickers, C.; Willcocks, D.; Farland, W. IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit. Rev. Toxicol. 2006, 36, 781–792. [Google Scholar] [CrossRef]
- Boobis, A.R.; Doe, J.E.; Heinrich-Hirsch, B.; Meek, M.E.; Munn, S.; Ruchirawat, M.; Schlatter, J.; Seed, J.; Vickers, C. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit. Rev. Toxicol. 2008, 38, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Ellwein, L.B. Proliferative and genotoxic cellular effects in 2-acetylaminofluorene bladder and liver carcinogenesis: Biological modeling of the ED01 study. Toxicol. Appl. Pharmacol. 1990, 104, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Murasaki, G.; Cohen, S.M. Co-carcinogenicity of sodium saccharin and N-[4-(5-nitro-2-furyl)-2-thiazolyl]formamide for the urinary bladder. Carcinogenesis 1983, 4, 97–99. [Google Scholar] [CrossRef]
- Cohen, S.M.; Boobis, A.R.; Meek, M.E.; Preston, R.J.; McGregor, D.B. 4-Aminobiphenyl and DNA reactivity: Case study within the context of the 2006 IPCS Human Relevance Framework for Analysis of a cancer mode of action for humans. Crit. Rev. Toxicol. 2006, 36, 803–819. [Google Scholar] [CrossRef]
- Ohnishi, T.; Arnold, L.L.; He, J.; Clark, N.M.; Kawasaki, S.; Rennard, S.I.; Boyer, C.W.; Cohen, S.M. Inhalation of tobacco smoke induces increased proliferation of urinary bladder epithelium and endothelium in female C57BL/6 mice. Toxicology 2007, 241, 58–65. [Google Scholar] [CrossRef]
- Hilton, G.M.; Adcock, C.; Akerman, G.; Baldassari, J.; Battalora, M.; Casey, W.; Clippinger, A.J.; Cope, R.; Goetz, A.; Hayes, A.W.; et al. Rethinking chronic toxicity and carcinogenicity assessment for agrochemicals project (ReCAAP): A reporting framework to support a weight of evidence safety assessment without long-term rodent bioassays. Regul. Toxicol. Pharmacol. 2022, 131, 105160. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Colacci, A.; Corvi, R.; Vaccari, M.; Aguila, M.C.; Corvaro, M.; Delrue, N.; Desaulniers, D.; Ertych, N.; Jacobs, A.; et al. Chemical carcinogen safety testing: OECD expert group international consensus on the development of an integrated approach for the testing and assessment of chemical non-genotoxic carcinogens. Arch. Toxicol. 2020, 94, 2899–2923. [Google Scholar] [CrossRef]
- Paparella, M.; Colacci, A.; Jacobs, M.N. Uncertainties of testing methods: What do we (want to) know about carcinogenicity? Altex 2017, 34, 235–252. [Google Scholar] [CrossRef]
- OECD. Overview of Concepts and Available Guidance Related to Integrated Approaches to Testing and Assessment (IATA); OECD Series on Testing and Assessment, No 329; OECD: Paris, France, 2020. [Google Scholar]
- Wood, C.E.; Hukkanen, R.R.; Sura, R.; Jacobson-Kram, D.; Nolte, T.; Odin, M.; Cohen, S.M. Scientific and Regulatory Policy Committee (SRPC) Review: Interpretation and use of cell proliferation data in cancer risk assessment. Toxicol. Pathol. 2015, 43, 760–775. [Google Scholar] [CrossRef]
- Nolte, T.; Kaufmann, W.; Schorsch, F.; Soames, T.; Weber, E. Standardized assessment of cell proliferation: The approach of the RITA-CEPA working group. Exp. Toxicol. Pathol. 2005, 57, 91–103. [Google Scholar] [CrossRef]
- Yusof, Y.A.; Edwards, A.M. Stimulation of DNA synthesis in primary rat hepatocyte cultures by liver tumor promoters: Interactions with other growth factors. Carcinogenesis 1990, 11, 761–770. [Google Scholar] [CrossRef]
- Yamamoto, N.; Imazato, K.; Masumoto, A. Growth stimulation of adult rat hepatocytes in a primary culture by soluble factor(s) secreted from nonparenchymal liver cell. Cell Struct. Funct. 1989, 14, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Kikumoto, H.; Lake, B.G.; Kawamura, S. Lack of effect of metofluthrin and sodium phenobarbital on replicative DNA synthesis and Ki-67 mRNA expression in cultured human hepatocytes. Toxicol. Res. 2015, 4, 901–913. [Google Scholar] [CrossRef]
- Remnant, L.; Kochanova, N.Y.; Reid, C.; Cisneros-Soberanis, F.; Earnshaw, W.C. The intrinsically disorderly story of Ki-67. Open Biol. 2021, 11, 210120. [Google Scholar] [CrossRef] [PubMed]
- Uxa, S.; Castillo-Binder, P.; Kohler, R.; Stangner, K.; Muller, G.A.; Engeland, K. Ki-67 gene expression. Cell Death Differ. 2021, 28, 3357–3370. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Woods, A.L.; Levison, D.A. The assessment of cellular proliferation by immunohistochemistry: A review of currently available methods and their applications. Histochem. J. 1992, 24, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Muskhelishvili, L.; Latendresse, J.R.; Kodell, R.L.; Henderson, E.B. Evaluation of cell proliferation in rat tissues with BrdU, PCNA, Ki-67(MIB-5) immunohistochemistry and in situ hybridization for histone mRNA. J. Histochem. Cytochem. 2003, 51, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Friedewald, W.F.; Rous, P. The initiating and promoting elements in tumor production: An analysis of the effects of tar, benzpyrene, and methylcholanthrene on rabbit skin. J. Exp. Med. 1944, 80, 101–126. [Google Scholar] [CrossRef]
- Gordon, E.; Cohen, S.M.; Singh, P. Folpet-induced short term cytotoxic and proliferative changes in the mouse duodenum. Toxicol. Mech. Methods 2012, 22, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.M.; Wolf, J.C.; McCoy, A.; Suh, M.; Proctor, D.M.; Kirman, C.R.; Haws, L.C.; Harris, M.A. Comparison of toxicity and recovery in the duodenum of B6C3F1 mice following treatment with intestinal carcinogens captan, folpet, and hexavalent chromium. Toxicol. Pathol. 2017, 45, 1091–1101. [Google Scholar] [CrossRef]
- Andersen, M.E.; Meek, M.E.; Boorman, G.A.; Brusick, D.J.; Cohen, S.M.; Dragan, Y.P.; Frederick, C.B.; Goodman, J.I.; Hard, G.C.; O’Flaherty, E.J.; et al. Lessons learned in applying the U.S. EPA proposed cancer guidelines to specific compounds. Toxicol. Sci. 2000, 53, 159–172. [Google Scholar] [CrossRef]
- Cohen, S.M. Screening for human urinary bladder carcinogens: Two-year bioassay is unnecessary. Toxicol. Res. 2018, 7, 565–575. [Google Scholar] [CrossRef]
- Peffer, R.C.; LeBaron, M.J.; Battalora, M.; Bomann, W.H.; Werner, C.; Aggarwal, M.; Rowe, R.R.; Tinwell, H. Minimum datasets to establish a CAR-mediated mode of action for rodent liver tumors. Regul. Toxicol. Pharmacol. 2018, 96, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Strupp, C.; Quesnot, N.; Richert, L.; Moore, J.; Bomann, W.H.; Singh, P. Weight of evidence and human relevance evaluation of the benfluralin mode of action in rodents (Part I): Liver carcinogenesis. Regul. Toxicol. Pharmacol. 2020, 117, 104758. [Google Scholar] [CrossRef]
- Strupp, C.; Bomann, W.H.; Spezia, F.; Gervais, F.; Forster, R.; Richert, L.; Singh, P. A human relevance investigation of PPARalpha-mediated key events in the hepatocarcinogenic mode of action of propaquizafop in rats. Regul. Toxicol. Pharmacol. 2018, 95, 348–361. [Google Scholar] [CrossRef]
- Soldatow, V.; Peffer, R.C.; Trask, O.J.; Cowie, D.E.; Andersen, M.E.; LeCluyse, E.; Deisenroth, C. Development of an in vitro high content imaging assay for quantitative assessment of CAR-dependent mouse, rat, and human primary hepatocyte proliferation. Toxicol. In Vitro 2016, 36, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Dellarco, V.L.; McGregor, D.; Berry, S.C.; Cohen, S.M.; Boobis, A.R. Thiazopyr and thyroid disruption: Case study within the context of the 2006 IPCS Human Relevance Framework for analysis of a cancer mode of action. Crit. Rev. Toxicol. 2006, 36, 793–801. [Google Scholar] [CrossRef]
- Cruzan, G.; Bus, J.; Banton, M.; Gingell, R.; Carlson, G. Mouse specific lung tumors from CYP2F2-mediated cytotoxic metabolism: An endpoint/toxic response where data from multiple chemicals converge to support a mode of action. Regul. Toxicol. Pharmacol. 2009, 55, 205–218. [Google Scholar] [CrossRef]
- Strupp, C.; Banas, D.A.; Cohen, S.M.; Gordon, E.B.; Jaeger, M.; Weber, K. Relationship of metabolism and cell proliferation to the mode of action of fluensulfone-induced mouse lung tumors: Analysis of their human relevance using the IPCS framework. Toxicol. Sci. 2012, 128, 284–294. [Google Scholar] [CrossRef]
- Strupp, C.; Bomann, W.; Cohen, S.M.; Weber, K. Relationship of metabolism and cell proliferation to the mode of action of fluensulfone-induced mouse lung tumors. II: Additional mechanistic studies. Toxicol. Sci. 2016, 154, 296–308. [Google Scholar] [CrossRef]
- Yoshida, Y.; Tatematsu, M.; Takaba, K.; Iwasaki, S.; Ito, N. Target organ specificity of cell proliferation induced by various carcinogens. Toxicol. Pathol. 1993, 21, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Umemura, T.; Okazaki, K.; Kanki, K.; Imazawa, T.; Masegi, T.; Nishikawa, A.; Hirose, M. Enhancing effects of simultaneous treatment with sodium nitrite on 2-amino-3-methylimidazo[4,5-f]quinoline-induced rat liver, colon and Zymbal’s gland carcinogenesis after initiation with diethylnitrosamine and 1,2-dimethylhydrazine. Int. J. Cancer 2006, 118, 2399–2404. [Google Scholar] [CrossRef]
- Suzuki, S.; Arnold, L.L.; Ohnishi, T.; Cohen, S.M. Effects of inorganic arsenic on the rat and mouse urinary bladder. Toxicol. Sci. 2008, 106, 350–363. [Google Scholar] [CrossRef]
- Corton, J.C. (US Environmental Protection Agency, Washington, DC, USA). Unpublished work. 2023. [Google Scholar]
- Parmentier, C.; Couttet, P.; Uteng, M.; Wolf, A.; Richert, L. Transcriptomic analysis of cholestatic compounds in vitro. Methods Mol. Biol. 2019, 1981, 175–186. [Google Scholar] [PubMed]
- Lauschke, V.M.; Hendriks, D.F.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D culture systems for studies of human liver function and assessments of the hepatotoxicity of drugs and drug candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef] [PubMed]
- Hopperstad, K.; Deisenroth, C. Development of a bioprinter-based method for incorporating metabolic competence into high-throughput in vitro assays. Front. Toxicol. 2023, 5, 1196245. [Google Scholar] [CrossRef]
- Baze, A.; Parmentier, C.; Hendriks, D.F.G.; Hurrell, T.; Heyd, B.; Bachellier, P.; Schuster, C.; Ingelman-Sundberg, M.; Richert, L. Three-dimensional spheroid primary human hepatocytes in monoculture and coculture with nonparenchymal cells. Tissue Eng. Part. C Methods 2018, 24, 534–545. [Google Scholar] [CrossRef]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, A.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human liver spheroids as a model to study aetiology and treatment of hepatic fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef]
- OECD. Guidance Document on the Characterisation, Validation and Reporting of Physiologically Based Kinetic (PBK) Models for Regulatory Purposes; OECD Series on Testing and Assessment No. 331; OECD: Paris, France, 2021. [Google Scholar]
- Ames, B.N.; Gold, L.S. Too many rodent carcinogens: Mitogenesis increases mutagenesis. Science 1990, 249, 970–971. [Google Scholar] [CrossRef]
- Cohen, S.M. The relevance of experimental carcinogenicity studies to human safety. Curr. Opin. Toxicol. 2017, 3, 6–11. [Google Scholar] [CrossRef]
- Hoffmann, B.; Piasecki, A.; Paul, D. Proliferation of fetal rat hepatocytes in response to growth factors and hormones in primary culture. J. Cell. Physiol. 1989, 139, 654–662. [Google Scholar] [CrossRef]
- Mitaka, T.; Sattler, G.L.; Pitot, H.C. Amino acid-rich medium (Leibovitz L-15) enhances and prolongs proliferation of primary cultured rat hepatocytes in the absence of serum. J. Cell. Physiol. 1991, 147, 495–504. [Google Scholar] [CrossRef]
- Plant, N.J.; Horley, N.J.; Dickins, M.; Hasmall, S.; Elcombe, C.R.; Bell, D.R. The coordinate regulation of DNA synthesis and suppression of apoptosis is differentially regulated by the liver growth agents, phenobarbital and methylclofenapate. Carcinogenesis 1998, 19, 1521–1527. [Google Scholar] [CrossRef]
- Enat, R.; Jefferson, D.M.; Ruiz-Opazo, N.; Gatmaitan, Z.; Leinwand, L.A.; Reid, L.M. Hepatocyte proliferation in vitro: Its dependence on the use of serum-free hormonally defined medium and substrata of extracellular matrix. Proc. Natl. Acad. Sci. USA 1984, 81, 1411–1415. [Google Scholar] [CrossRef]
- Hirose, Y.; Nagahori, H.; Yamada, T.; Deguchi, Y.; Tomigahara, Y.; Nishioka, K.; Uwagawa, S.; Kawamura, S.; Isobe, N.; Lake, B.G.; et al. Comparison of the effects of the synthetic pyrethroid Metofluthrin and phenobarbital on CYP2B form induction and replicative DNA synthesis in cultured rat and human hepatocytes. Toxicology 2009, 258, 64–69. [Google Scholar] [CrossRef]
- Parzefall, W.; Erber, E.; Sedivy, R.; Schulte-Hermann, R. Testing for induction of DNA synthesis in human hepatocyte primary cultures by rat liver tumor promoters. Cancer Res. 1991, 51, 1143–1147. [Google Scholar]
- LaRocca, J.L.; Rasoulpour, R.J.; Gollapudi, B.B.; Eisenbrandt, D.L.; Murphy, L.A.; LeBaron, M.J. Integration of novel approaches demonstrates simultaneous metabolic inactivation and CAR-mediated hepatocarcinogenesis of a nitrification inhibitor. Toxicol. Rep. 2017, 4, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Breier, J.M.; Radio, N.M.; Mundy, W.R.; Shafer, T.J. Development of a high-throughput screening assay for chemical effects on proliferation and viability of immortalized human neural progenitor cells. Toxicol. Sci. 2008, 105, 119–133. [Google Scholar] [CrossRef]
- Thermofischer Scientific. Click-iT® EdU HCS Assays Protocol. Available online: https://www.thermofisher.com/in/en/home/references/protocols/cell-and-tissue-analysis/protocols/click-it-edu-hcs-assays-protocol.html (accessed on 10 August 2023).
- Carnahan, J.; Beltran, P.J.; Babij, C.; Le, Q.; Rose, M.J.; Vonderfecht, S.; Kim, J.L.; Smith, A.L.; Nagapudi, K.; Broome, M.A.; et al. Selective and potent Raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol. Cancer Ther. 2010, 9, 2399–2410. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Onda, M.; Kim, C.; Xiang, L.; Weldon, J.E.; Lee, B.; Pastan, I. A recombinant immunotoxin engineered for increased stability by adding a disulfide bond has decreased immunogenicity. Protein Eng. Des. Sel. 2012, 25, 1–6. [Google Scholar] [CrossRef]
- Narazaki, M.; Segarra, M.; Hou, X.; Tanaka, T.; Li, X.; Tosato, G. Oligo-guanosine nucleotide induces neuropilin-1 internalization in endothelial cells and inhibits angiogenesis. Blood 2010, 116, 3099–3107. [Google Scholar] [CrossRef]
- Kangas, L.; Grönroos, M.; Nieminen, A.L. Bioluminescence of cellular ATP: A new method for evaluating cytotoxic agents in vitro. Med. Biol. 1984, 62, 338–343. [Google Scholar] [PubMed]
- Barltrop, J.A.; Owen, T.C.; Cory, A.H.; Cory, J.G. 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl)tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans as cell-viability indicators. Bioorganic Med. Chem. Lett. 1991, 1, 611–614. [Google Scholar]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A. Comparison of MTT, XTT, and a novel tetrazolium compound MTS for in vitro proliferation and chemosensitivity assays. Mol. Biol. Cell 1992, 3, 184A. [Google Scholar]
- Giuliano, K.; Gough, A.; Taylor, D.; Vernetti, L.; Johnston, P. Early safety assessment using cellular systems biology yields insights into mechanisms of action. J. Biomol. Screen. 2010, 15, 783–797. [Google Scholar] [CrossRef]
- Knight, A.W.; Little, S.; Houck, K.; Dix, D.; Judson, R.; Richard, A.; McCarroll, N.; Akerman, G.; Yang, C.; Birrell, L.; et al. Evaluation of high-throughput genotoxicity assays used in profiling the US EPA ToxCast chemicals. Regul. Toxicol. Pharmacol. 2009, 55, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Holsapple, M.P.; Pitot, H.C.; Cohen, S.M.; Boobis, A.R.; Klaunig, J.E.; Pastoor, T.; Dellarco, V.L.; Dragan, Y.P. Mode of action in relevance of rodent liver tumors to human cancer risk. Toxicol. Sci. 2006, 89, 51–56. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Babich, M.A.; Baetcke, K.P.; Cook, J.C.; Corton, J.C.; David, R.M.; DeLuca, J.G.; Lai, D.Y.; McKee, R.H.; Peters, J.M.; et al. PPARalpha agonist-induced rodent tumors: Modes of action and human relevance. Crit. Rev. Toxicol. 2003, 33, 655–780. [Google Scholar] [CrossRef]
- Laine, L.; Ahnen, D.; McClain, C.; Solcia, E.; Walsh, J.H. Review article: Potential gastrointestinal effects of long-term acid suppression with proton pump inhibitors. Aliment. Pharmacol. Ther. 2000, 14, 651–668. [Google Scholar] [CrossRef]
- Cohen, S.M.; Gordon, E.B.; Singh, P.; Arce, G.T.; Nyska, A. Carcinogenic mode of action of folpet in mice and evaluation of its relevance to humans. Crit. Rev. Toxicol. 2010, 40, 531–545. [Google Scholar] [CrossRef]
- Bhat, V.S.; Cohen, S.M.; Gordon, E.B.; Wood, C.E.; Cullen, J.M.; Harris, M.A.; Proctor, D.M.; Thompson, C.M. An adverse outcome pathway for small intestinal tumors in mice involving chronic cytotoxicity and regenerative hyperplasia: A case study with hexavalent chromium, captan, and folpet. Crit. Rev. Toxicol. 2020, 50, 685–706. [Google Scholar] [CrossRef]
- GPO. Federal Register. In Information; U.S. Government Publishing Office: Washington, DC, USA, 2004; Volume 69. [Google Scholar]
- IARC. Species Differences in Thyroid, Kidney and Urinary Bladder Carcinogenesis; IARC: Lyon, France, 1999; pp. 1–32. [Google Scholar]
- NTP. 15th Report on Carcinogens; National Toxicology Program Department of Health and Human Services, Public Health Service: Washington, DC, USA, 2021. [Google Scholar]
- Van der Laan, J.W.; Kasper, P.; Silva Lima, B.; Jones, D.R.; Pasanen, M. Critical analysis of carcinogenicity study outcomes. Relationship with pharmacological properties. Crit. Rev. Toxicol. 2016, 46, 587–614. [Google Scholar] [CrossRef]
- WHO. Interpretation of negative epidemiological evidence for carcinogenicity. In Proceedings of the Symposium, Oxford, UK, 4–6 July 1983; pp. 1–232.
- Cohen, S.M.; Zhongyu, Y.; Bus, J.S. Relevance of mouse lung tumors to human risk assessment. J. Toxicol. Environ. Health B Crit. Rev. 2020, 23, 214–241. [Google Scholar] [CrossRef]
- Desaulniers, D.; Vasseur, P.; Jacobs, A.; Aguila, M.C.; Ertych, N.; Jacobs, M.N. Integration of epigenetic mechanisms into non-genotoxic carcinogenicity hazard assessment: Focus on DNA methylation and histone modifications. Int. J. Mol. Sci. 2021, 22, 10969. [Google Scholar] [CrossRef]
- Oku, Y.; Madia, F.; Lau, P.; Paparella, M.; McGovern, T.; Luijten, M.; Jacobs, M.N. Analyses of transcriptomics cell signalling for pre-screening applications in the integrated approach for testing and assessment of non-genotoxic carcinogens. Int. J. Mol. Sci. 2022, 23, 12718. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Fountzilas, E.; Bleris, L.; Kurzrock, R. Transcriptomics and solid tumors: The next frontier in precision cancer medicine. Semin. Cancer Biol. 2022, 84, 50–59. [Google Scholar] [CrossRef]
- Khalighi, S.; Singh, S.; Varadan, V. Untangling a complex web: Computational analyses of tumor molecular profiles to decode driver mechanisms. J. Genet. Genom. 2020, 47, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Yauk, C.L.; Harrill, A.H.; Ellinger-Ziegelbauer, H.; van der Laan, J.W.; Moggs, J.; Froetschl, R.; Sistare, F.; Pettit, S. A cross-sector call to improve carcinogenicity risk assessment through use of genomic methodologies. Regul. Toxicol. Pharmacol. 2020, 110, 104526. [Google Scholar] [CrossRef] [PubMed]
- Ahir, B.K.; Sanders, A.P.; Rager, J.E.; Fry, R.C. Systems biology and birth defects prevention: Blockade of the glucocorticoid receptor prevents arsenic-induced birth defects. Environ. Health Perspect. 2013, 121, 332–338. [Google Scholar] [CrossRef]
- Waters, M.D.; Jackson, M.; Lea, I. Characterizing and predicting carcinogenicity and mode of action using conventional and toxicogenomics methods. Mutat. Res. 2010, 705, 184–200. [Google Scholar] [CrossRef]
- Corton, J.C.; Kleinstreuer, N.C.; Judson, R.S. Identification of potential endocrine disrupting chemicals using gene expression biomarkers. Toxicol. Appl. Pharmacol. 2019, 380, 114683. [Google Scholar] [CrossRef]
- Corton, J.C.; Mitchell, C.A.; Auerbach, S.; Bushel, P.; Ellinger-Ziegelbauer, H.; Escobar, P.A.; Froetschl, R.; Harrill, A.H.; Johnson, K.; Klaunig, J.E.; et al. A collaborative initiative to establish genomic biomarkers for assessing tumorigenic potential to reduce reliance on conventional rodent carcinogenicity studies. Toxicol. Sci. 2022, 188, 4–16. [Google Scholar] [CrossRef]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.J.; Ma, D.; Liu, Y.Y.; Xiao, Y.; Gong, Y.; Jiang, Y.Z.; Shao, Z.M.; Hu, X.; Di, G.H. Bulk and single-cell transcriptome profiling reveal the metabolic heterogeneity in human breast cancers. Mol. Ther. 2021, 29, 2350–2365. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.P.; Oshida, K.; Kumar, R.; Baldwin, W.S.; Corton, J.C. Chemical activation of the constitutive androstane receptor leads to activation of oxidant-induced Nrf2. Toxicol. Sci. 2019, 167, 172–189. [Google Scholar] [CrossRef]
- Avila, A.M.; Bebenek, I.; Bonzo, J.A.; Bourcier, T.; Davis Bruno, K.L.; Carlson, D.B.; Dubinion, J.; Elayan, I.; Harrouk, W.; Lee, S.L.; et al. An FDA/CDER perspective on nonclinical testing strategies: Classical toxicology approaches and new approach methodologies (NAMs). Regul. Toxicol. Pharmacol. 2020, 114, 104662. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, G.; Derr, R.S.; Misovic, B.; Morolli, B.; Calleja, F.M.; Vrieling, H. The extended ToxTracker assay discriminates between induction of DNA damage, oxidative stress, and protein misfolding. Toxicol. Sci. 2016, 150, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.U.; Das, A.; Rogkoti, V.-M.; Fokkelman, M.; Marcotte, R.; de Jong, C.G.; Koedoot, E.; Lee, J.S.; Meilijson, I.; Hannenhalli, S. Migration rather than proliferation transcriptomic signatures are strongly associated with breast cancer patient survival. Sci. Rep. 2019, 9, 10989. [Google Scholar] [CrossRef]
- ICH. Testing for Carcinogenicity of Pharmaceuticals S1B(R1); ICH: Geneva, Switzerland, 2022. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ranking | Assay Title | Alias | Critical Aspects and Potential Limitations | Key References |
|---|---|---|---|---|
| Category A | Proliferation markers (in vivo) | BrdU or Ki-67 in vivo | It is critical that cell number is used as denominator and focused on the correct target cell | [5,32,33,64,65] |
| Category B/C | In vitro proliferation in primary cells | Serum-free liver mitogen test, CAR assay, BrdU in vitro | Metabolic competence should be characterized; high variability for human tissue; quality of preparation (e.g., presence of non-target cell types); issues regarding primary human cells (e.g., ethics, disease history, contaminants, viruses, number and sex of donors); reproducibility issues | [14,36,66,67,68,69,70,71,72] |
| In vitro proliferation in cell lines | DNA synthesis proliferation (BrdU in vitro, 14C-thymidine), BRAF inhibitors | Limited metabolic competence and basal proliferation should be taken into account; perturbation of cell signaling due to immortalization | [73,74,75,76,77] | |
| Category C | Assays addressing only cell number (or metabolic activity as a marker proportional to cell number) | CCK8, CellTiter-GloTM, CellTiter 96AQ, CellCiphr® Premier | Cytotoxicity assays, not specific to proliferation | [78,79,80,81,82,83,84,85,86] |
| Prototypical Activator | Target Organ | Mechanism | Comment | Key References |
|---|---|---|---|---|
| Phenobarbital | Liver, thyroid | Nuclear receptor binding (CAR), metabolic phase II induction, thyroid (T)-hormone clearance, and constant feedback stimulation | Found in rats and mice | [46,87] |
| Thiazopyr | Thyroid | Metabolic phase II induction, T-hormone clearance, and constant feedback stimulation | Found in rats | [50] |
| Clofibrate, Wyeth (WY)-14,643 | Liver, pancreas, testis | Nuclear receptor binding (PPARα), metabolic phase II induction, T-hormone clearance, and constant feedback stimulation | Found in rats and mice | [17,48,88] |
| Chloroform, methapyrilene | Liver, kidney | Cytotoxicity/repair | At cytotoxic doses | [13,44] |
| Omeprazol, chlorothalonil | Stomach, neuro-endocrine | Gastrin-induced mitogenesis | - | [89] |
| Folpet, chromium | Duodenum | Cytotoxicity/repair | At cytotoxic doses | [90,91,92] |
| D-Limonene, nitrapyrin | Kidney | Alpha 2u-globulin | Found in male rats | [93] |
| Sodium saccharin, ascorbate | Bladder | Crystal formation and chronic local irritation | Found in rats | [94] |
| Estrogen | Mammary gland | Constant mitogenicity | - | [94] |
| Cyclosporin A | Lymphoma, squamous cell carcinoma | Immunosuppression | Activation of viral carcinogens, not directly carcinogenic; carcinogenic in Xpa/p53 mice | [95] |
| Isoniazid | Lung | Mitogen | Found in mice | [96,97] |
| Fluensulfone, Styrene | Lung | Metabolic induction and resulting damage in Club cells | Found in mice | [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strupp, C.; Corvaro, M.; Cohen, S.M.; Corton, J.C.; Ogawa, K.; Richert, L.; Jacobs, M.N. Increased Cell Proliferation as a Key Event in Chemical Carcinogenesis: Application in an Integrated Approach for the Testing and Assessment of Non-Genotoxic Carcinogenesis. Int. J. Mol. Sci. 2023, 24, 13246. https://doi.org/10.3390/ijms241713246
Strupp C, Corvaro M, Cohen SM, Corton JC, Ogawa K, Richert L, Jacobs MN. Increased Cell Proliferation as a Key Event in Chemical Carcinogenesis: Application in an Integrated Approach for the Testing and Assessment of Non-Genotoxic Carcinogenesis. International Journal of Molecular Sciences. 2023; 24(17):13246. https://doi.org/10.3390/ijms241713246
Chicago/Turabian StyleStrupp, Christian, Marco Corvaro, Samuel M. Cohen, J. Christopher Corton, Kumiko Ogawa, Lysiane Richert, and Miriam N. Jacobs. 2023. "Increased Cell Proliferation as a Key Event in Chemical Carcinogenesis: Application in an Integrated Approach for the Testing and Assessment of Non-Genotoxic Carcinogenesis" International Journal of Molecular Sciences 24, no. 17: 13246. https://doi.org/10.3390/ijms241713246