
KCNG4 Genetic Variant Linked to Migraine Prevents Expression of KCNB1




Abstract
:1. Introduction
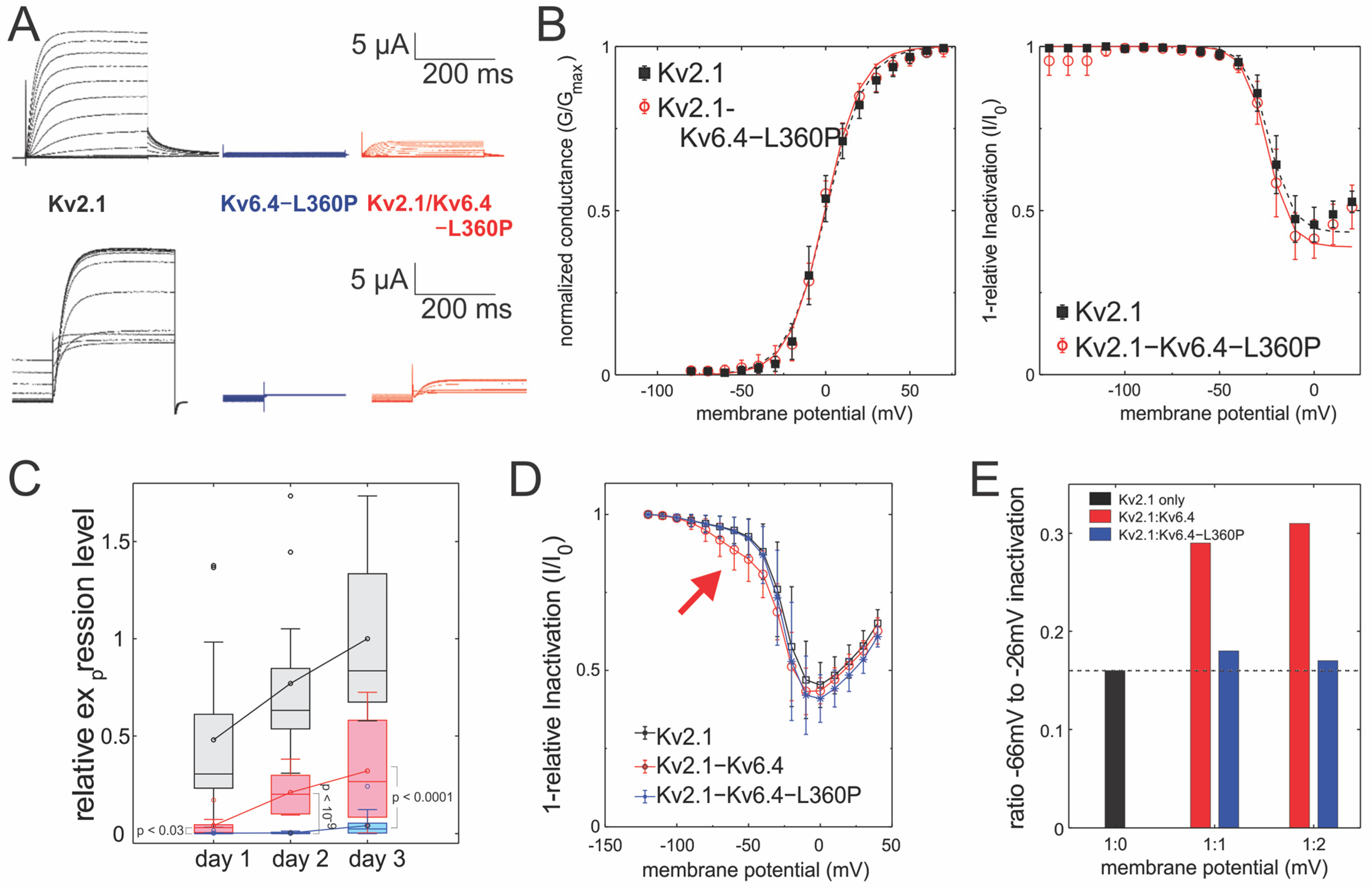
2. Results
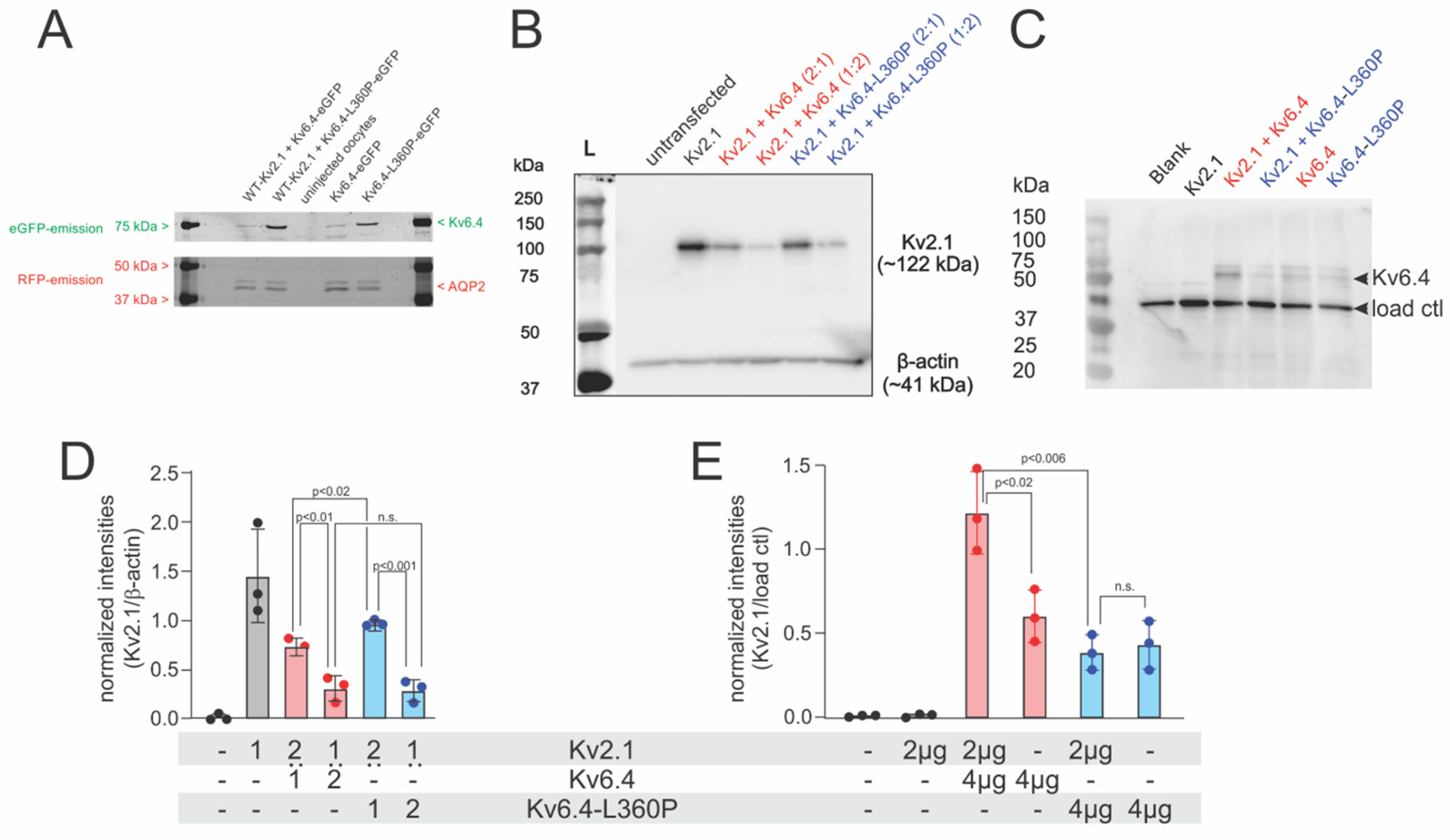
2.1. Kv2.1/Kv6.4-L360P Complexes Are Strongly Translated and Assembled
2.2. Kv2.1/Kv6.4-L360P Complexes Are Not Trafficked to the Plasma Membrane
3. Discussion
4. Materials and Methods
4.1. Pulse Protocols and Data Analysis
4.2. Expression of Kv2/Kv6 in Xenopus Oocytes
4.3. Immunoblotting of Kv2.1 and Kv6.4
4.3.1. Xenopus Oocytes
4.3.2. Mammalian Cell Lines
4.3.3. Cell Surface Biotinylation Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hille, B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J. 1978, 22, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Bar, L.; Shalom, L.; Lezmy, J.; Peretz, A.; Attali, B. Excitatory and inhibitory hippocampal neurons differ in their homeostatic adaptation to chronic M-channel modulation. Front. Mol. Neurosci. 2022, 15, 972023. [Google Scholar] [CrossRef]
- Piccialli, I.; Sisalli, M.J.; de Rosa, V.; Boscia, F.; Tedeschi, V.; Secondo, A.; Pannaccione, A. Increased KV2.1 Channel Clustering Underlies the Reduction of Delayed Rectifier K+ Currents in Hippocampal Neurons of the Tg2576 Alzheimer’s Disease Mouse. Cells 2022, 11, 2820. [Google Scholar] [CrossRef]
- Ottschytsch, N.; Raes, A.; Van Hoorick, D.; Snyders, D.J. Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome. Proc. Natl. Acad. Sci. USA 2002, 99, 7986–7991. [Google Scholar] [CrossRef]
- Shen, N.V.; Chen, X.; Boyer, M.M.; Pfaffinger, P.J. Deletion analysis of K+ channel assembly. Neuron 1993, 11, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Isacoff, E.Y.; Jan, Y.N.; Jan, L.Y. Evidence for the formation of heteromultimeric potassium channels in Xenopus oocytes. Nature 1990, 345, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Bocksteins, E. Kv5, Kv6, Kv8, and Kv9 subunits: No simple silent bystanders. J. Gen. Physiol. 2016, 147, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Bocksteins, E.; Snyders, D.J. Electrically silent Kv subunits: Their molecular and functional characteristics. Physiology 2012, 27, 73–84. [Google Scholar] [CrossRef]
- David, J.P.; Stas, J.I.; Schmitt, N.; Bocksteins, E. Auxiliary KCNE subunits modulate both homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4 channels. Sci. Rep. 2015, 5, 12813. [Google Scholar] [CrossRef]
- Bocksteins, E.; Labro, A.J.; Mayeur, E.; Bruyns, T.; Timmermans, J.P.; Adriaensen, D.; Snyders, D.J. Conserved negative charges in the N-terminal tetramerization domain mediate efficient assembly of Kv2.1 and Kv2.1/Kv6.4 channels. J. Biol. Chem. 2009, 284, 31625–31634. [Google Scholar] [CrossRef]
- Bocksteins, E.; Labro, A.J.; Snyders, D.J.; Mohapatra, D.P. The electrically silent Kv6.4 subunit confers hyperpolarized gating charge movement in Kv2.1/Kv6.4 heterotetrameric channels. PLoS ONE 2012, 7, e37143. [Google Scholar] [CrossRef]
- Bocksteins, E.; Mayeur, E.; Van Tilborg, A.; Regnier, G.; Timmermans, J.P.; Snyders, D.J. The subfamily-specific interaction between Kv2.1 and Kv6.4 subunits is determined by interactions between the N- and C-termini. PLoS ONE 2014, 9, e98960. [Google Scholar] [CrossRef]
- Möller, L.; Regnier, G.; Labro, A.J.; Blunck, R.; Snyders, D.J. Determining the correct stoichiometry of Kv2.1/Kv6.4 heterotetramers, functional in multiple stoichiometrical configurations. Proc. Natl. Acad. Sci. USA 2020, 117, 9365–9376. [Google Scholar] [CrossRef] [PubMed]
- Lafrenière, R.G.; Rouleau, G.A. Identification of novel genes involved in migraine. Headache 2012, 52 (Suppl. S2), 107–110. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Christensen, S.L.; Al-Karagholi, M.A.-M.; Olesen, J. Molecular nociceptive mechanisms in migraine: The migraine cascade. Eur. J. Neurol. 2024, 31, e16333. [Google Scholar] [CrossRef]
- Tsantoulas, C.; McMahon, S.B. Opening paths to novel analgesics: The role of potassium channels in chronic pain. Trends Neurosci. 2014, 37, 146–158. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Fernandez-Marino, A.I.; Tan, X.F.; Bae, C.; Huffer, K.; Jiang, J.; Swartz, K.J. Inactivation of the Kv2.1 channel through electromechanical coupling. Nature 2023, 622, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Haddad, G.A.; Blunck, R. Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J. Gen. Physiol. 2011, 137, 455–472. [Google Scholar] [CrossRef] [PubMed]
- von Heijne, G. Proline kinks in transmembrane alpha-helices. J. Mol. Biol. 1991, 218, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Rousseau, J.; Michaud, C.; Lourenco, C.M.; Stoler, J.M.; Louie, R.J.; Clarkson, L.K.; Lichty, A.; Koboldt, D.C.; Reshmi, S.C.; et al. Mono-allelic KCNB2 variants lead to a neurodevelopmental syndrome caused by altered channel inactivation. Am. J. Hum. Genet. 2024, 111, 761–777. [Google Scholar] [CrossRef]
- Hart, N.S.; Mountford, J.K.; Voigt, V.; Fuller-Carter, P.; Barth, M.; Nerbonne, J.M.; Hunt, D.M.; Carvalho, L.S. The Role of the Voltage-Gated Potassium Channel Proteins Kv8.2 and Kv2.1 in Vision and Retinal Disease: Insights from the Study of Mouse Gene Knock-Out Mutations. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Allen, N.M.; Weckhuysen, S.; Gorman, K.; King, M.D.; Lerche, H. Genetic potassium channel-associated epilepsies: Clinical review of the Kv family. Eur. J. Paediatr. Neurol. 2020, 24, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Combi, R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front. Cell. Neurosci. 2016, 10, 81. [Google Scholar] [CrossRef]
- Lee, M.C.; Nahorski, M.S.; Hockley, J.R.F.; Lu, V.B.; Ison, G.; Pattison, L.A.; Callejo, G.; Stouffer, K.; Fletcher, E.; Brown, C.; et al. Human Labor Pain Is Influenced by the Voltage-Gated Potassium Channel KV6.4 Subunit. Cell Rep. 2020, 32, 107941. [Google Scholar] [CrossRef]
- Crosson, T.; Bhat, S.; Wang, J.-C.; Salaun, C.; Roversi, K.; Herzog, H.; Rafei, M.; Blunck, R.; Talbot, S. IL-13 promotes sensory-sympathetic neurons crosstalk in asthma. bioRxiv 2023. [Google Scholar] [CrossRef]
- Messlinger, K.; Russo, A.F. Current understanding of trigeminal ganglion structure and function in headache. Cephalalgia 2019, 39, 1661–1674. [Google Scholar] [CrossRef]
- Yang, L.; Xu, M.; Bhuiyan, S.A.; Li, J.; Zhao, J.; Cohrs, R.J.; Susterich, J.T.; Signorelli, S.; Green, U.; Stone, J.R.; et al. Human and mouse trigeminal ganglia cell atlas implicates multiple cell types in migraine. Neuron 2022, 110, 1806–1821.e8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, F.; Zhang, Y.Y.; Lin, J.; Li, Y.L.; Zhou, C.; Li, C.J.; Shen, J.F. C-X-C motif chemokine ligand 1 and its receptor C-X-C motif chemokine receptor 2 in trigeminal ganglion contribute to nerve injury-induced orofacial mechanical allodynia. J. Oral Rehabil. 2022, 49, 195–206. [Google Scholar] [CrossRef]
- Tsantoulas, C. Emerging potassium channel targets for the treatment of pain. Curr. Opin. Support. Palliat. Care 2015, 9, 147–154. [Google Scholar] [CrossRef]
- Takeda, M.; Tsuboi, Y.; Kitagawa, J.; Nakagawa, K.; Iwata, K.; Matsumoto, S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol. Pain 2011, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Dussor, G.; Cao, Y.Q. TRPM8 and Migraine. Headache 2016, 56, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- LaPaglia, D.M.; Sapio, M.R.; Burbelo, P.D.; Thierry-Mieg, J.; Thierry-Mieg, D.; Raithel, S.J.; Ramsden, C.E.; Iadarola, M.J.; Mannes, A.J. RNA-Seq investigations of human post-mortem trigeminal ganglia. Cephalalgia 2018, 38, 912–932. [Google Scholar] [CrossRef]
- Andres-Enguix, I.; Shang, L.; Stansfeld, P.J.; Morahan, J.M.; Sansom, M.S.P.; Lafrenière, R.G.; Roy, B.; Griffiths, L.R.; Rouleau, G.A.; Ebers, G.C.; et al. Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci. Rep. 2012, 2, 237. [Google Scholar] [CrossRef] [PubMed]
- Lafrenière, R.G.; Rouleau, G.A. Migraine: Role of the TRESK two-pore potassium channel. Int. J. Biochem. Cell Biol. 2011, 43, 1533–1536. [Google Scholar] [CrossRef]
- Tewari, D.; Sattler, C.; Benndorf, K. Functional properties of a disease mutation for migraine in Kv2.1/6.4 channels. bioRxiv 2024. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conductance | V50 (mV) | Slope (mV) | Apparent Charge | N | |
| Kv2.1 | 0.1 ± 0.8 | 10.9 ± 0.7 | 2.37 ± 0.14 | 6 | |
| Kv2.1/Kv6.4 | −9.1 ± 0.6 | 15.9 ± 0.5 | 1.63 ± 0.05 | 18 | |
| Kv2.1/Kv6.4-L360P | −0.4 ± 0.6 | 10.7 ± 0.6 | 2.41 ± 0.12 | 9 | |
| Inactivation | V50(mV) | Slope (mV) | Apparent Charge | Maximum Inactivation | N |
| Kv2.1 | −23.8 ± 0.4 | −6.0 ± 0.3 | −4.19 ± 0.21 | 0.58 ± 0.02 | 9 |
| Kv2.1/Kv6.4 | −60.7 ± 0.6 | −8.4 ± 0.4 | −3.07 ± 0.14 | 0.55 ± 0.02 | 11 |
| Kv2.1/Kv6.4-L360P | −24.7 ± 0.6 | −6.1 ± 0.5 | −4.24 ± 0.33 | 0.61 ± 0.02 | 6 |
| Incubation Time | Constructs | Expression (µA) | Ratio to Kv2.1 (%) | |
|---|---|---|---|---|
| 18–24 h | Kv2.1 | 33.2 ± 30.5 | - | |
| Kv2.1/Kv6.4 | 2.9 ± 3.6 | 8.7 | }p < 0.03 | |
| Kv2.1/Kv6.4-L360P | 0.2 ± 0.3 | 0.6 | ||
| 36–48 h | Kv2.1 | 53.3 ± 28.5 | - | |
| Kv2.1/Kv6.4 | 14.6 ± 6.9 | 27.4 | }p < 10−9 | |
| Kv2.1/Kv6.4-L360P | 0.2 ± 0.3 | 0.4 | ||
| 66–72 h | Kv2.1 | 69.2 ± 28.8 | - | |
| Kv2.1/Kv6.4 | 22.3 ± 18.4 | 32.2 | }p < 0.0001 | |
| Kv2.1/Kv6.4-L360P | 2.9 ± 4.0 | 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacroix, G.; Bhat, S.; Shafia, Z.; Blunck, R. KCNG4 Genetic Variant Linked to Migraine Prevents Expression of KCNB1. Int. J. Mol. Sci. 2024, 25, 8960. https://doi.org/10.3390/ijms25168960
Lacroix G, Bhat S, Shafia Z, Blunck R. KCNG4 Genetic Variant Linked to Migraine Prevents Expression of KCNB1. International Journal of Molecular Sciences. 2024; 25(16):8960. https://doi.org/10.3390/ijms25168960
Chicago/Turabian StyleLacroix, Gabriel, Shreyas Bhat, Zerghona Shafia, and Rikard Blunck. 2024. "KCNG4 Genetic Variant Linked to Migraine Prevents Expression of KCNB1" International Journal of Molecular Sciences 25, no. 16: 8960. https://doi.org/10.3390/ijms25168960