
Genetic Diversity, Selection Signatures, and Genome-Wide Association Study Identify Candidate Genes Related to Litter Size in Hu Sheep


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Principal Component Analysis and Genetic Diversity Estimation
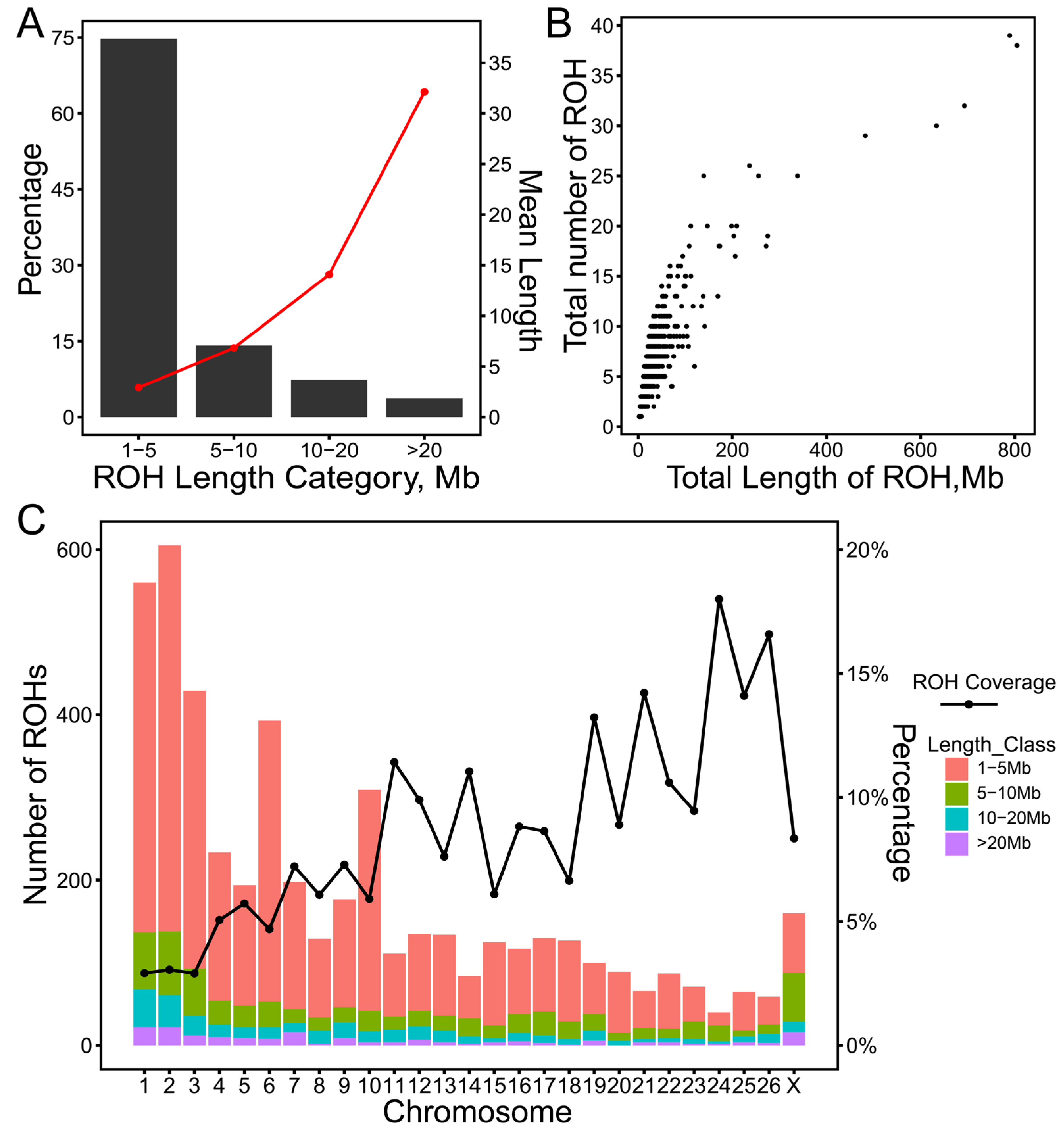
2.2. Distribution of Runs of Homozygosity
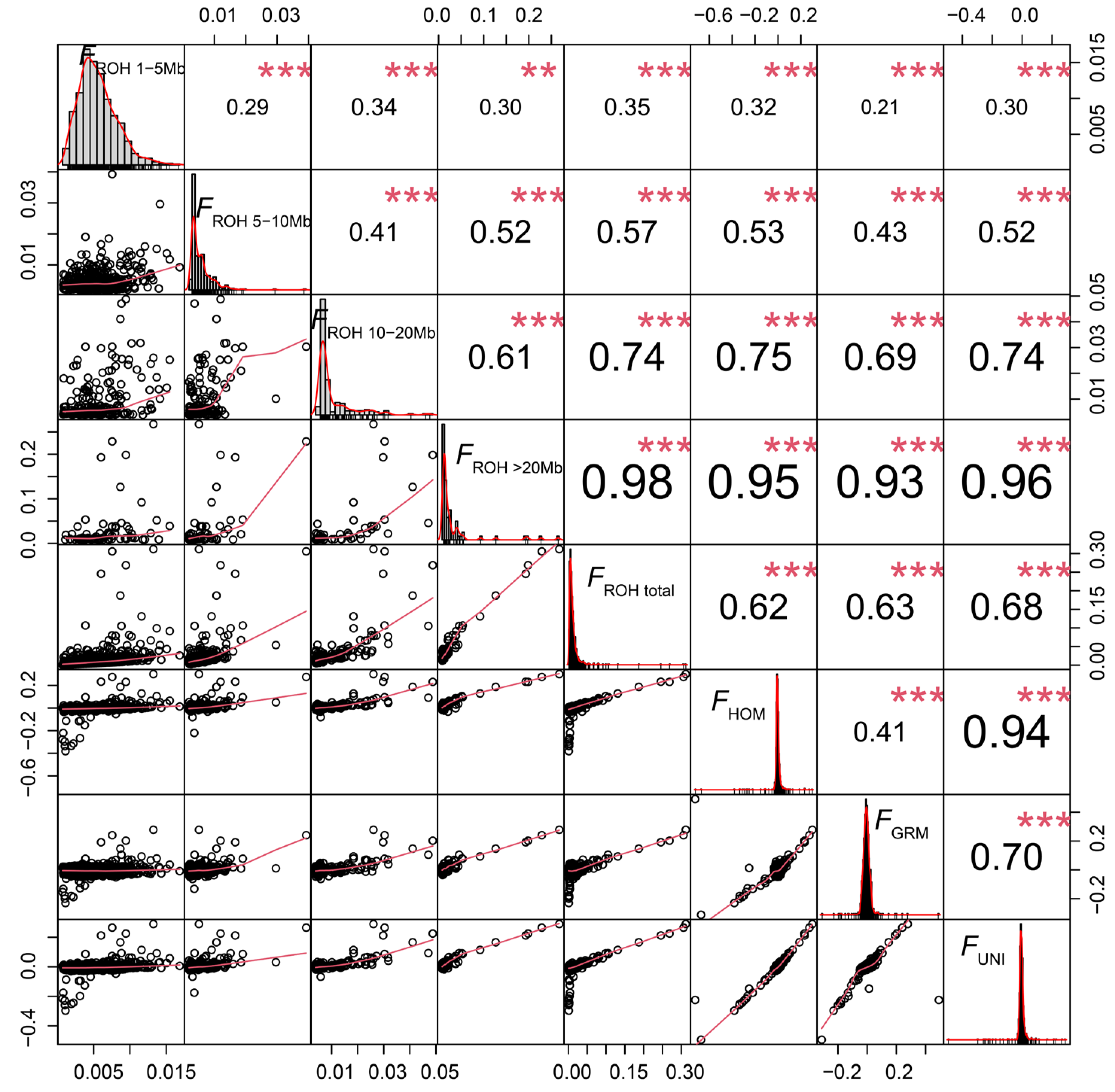
2.3. Estimation of Inbreeding Coefficients
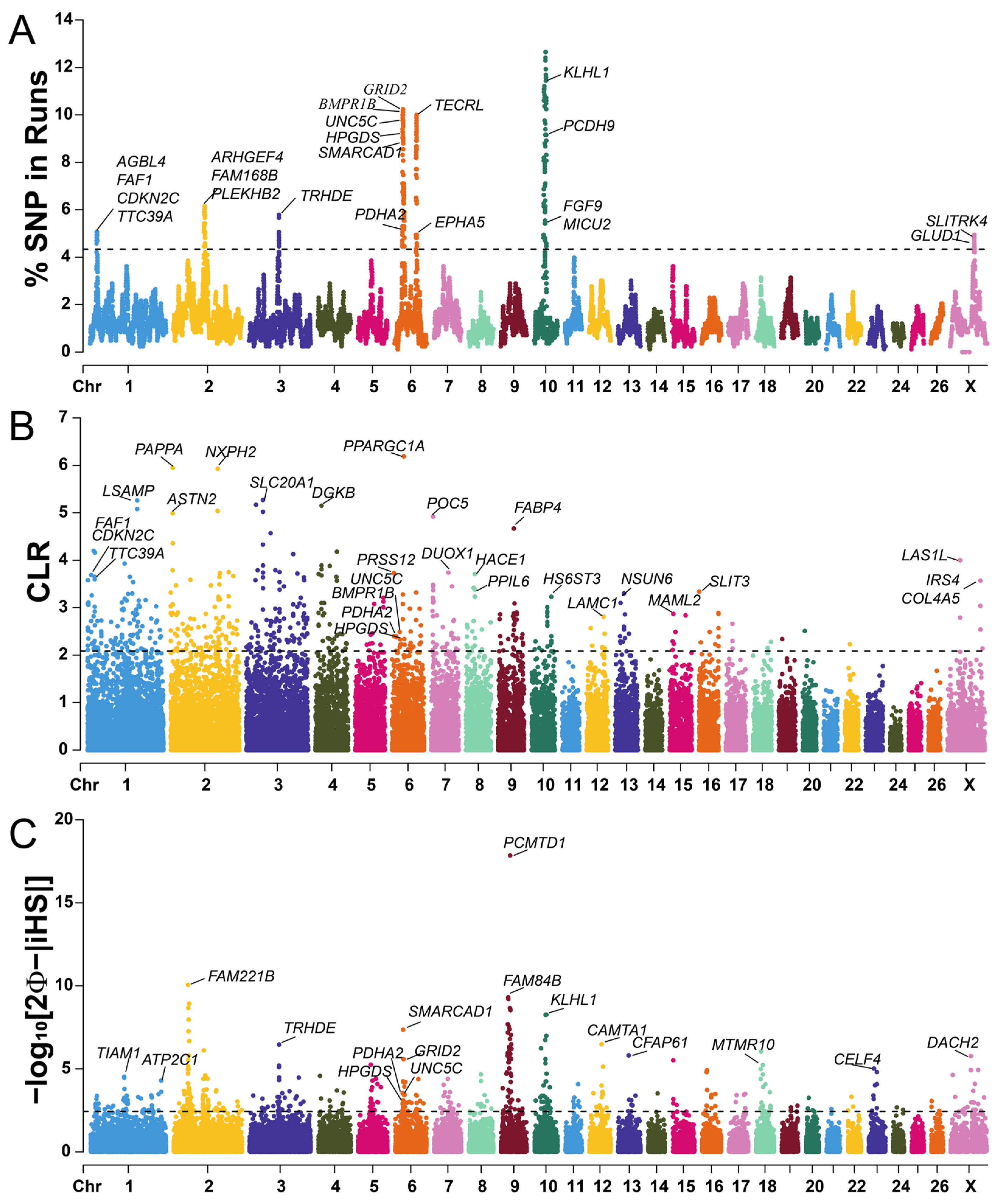
2.4. Detection of Selection Signatures
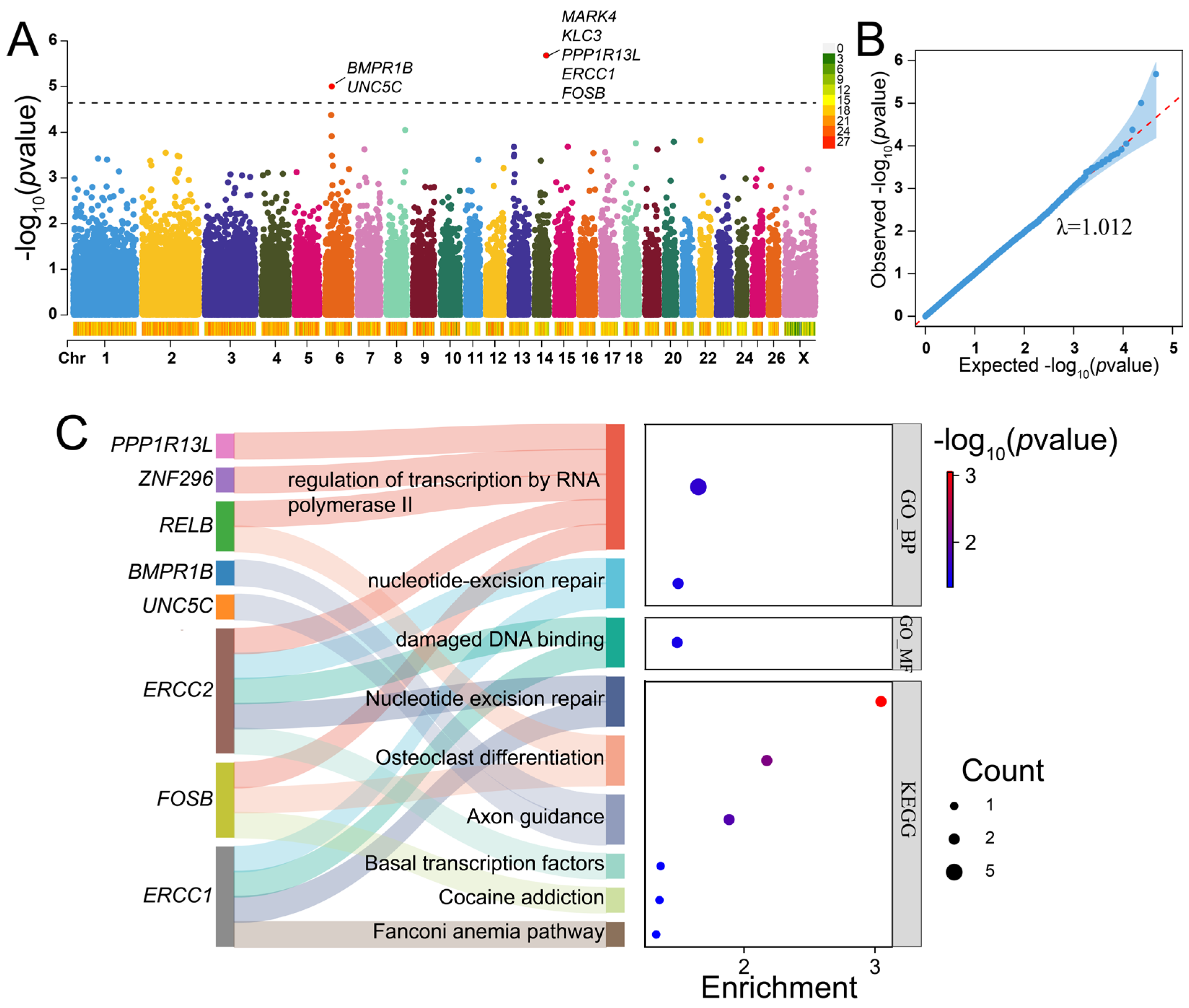
2.5. GWAS for Litter Size of Hu Sheep
3. Discussion
3.1. PCA and Genetic Diversity Analysis
3.2. Characteristics of Runs of Homozygosity
3.3. Genomic Inbreeding Coefficients Estimation
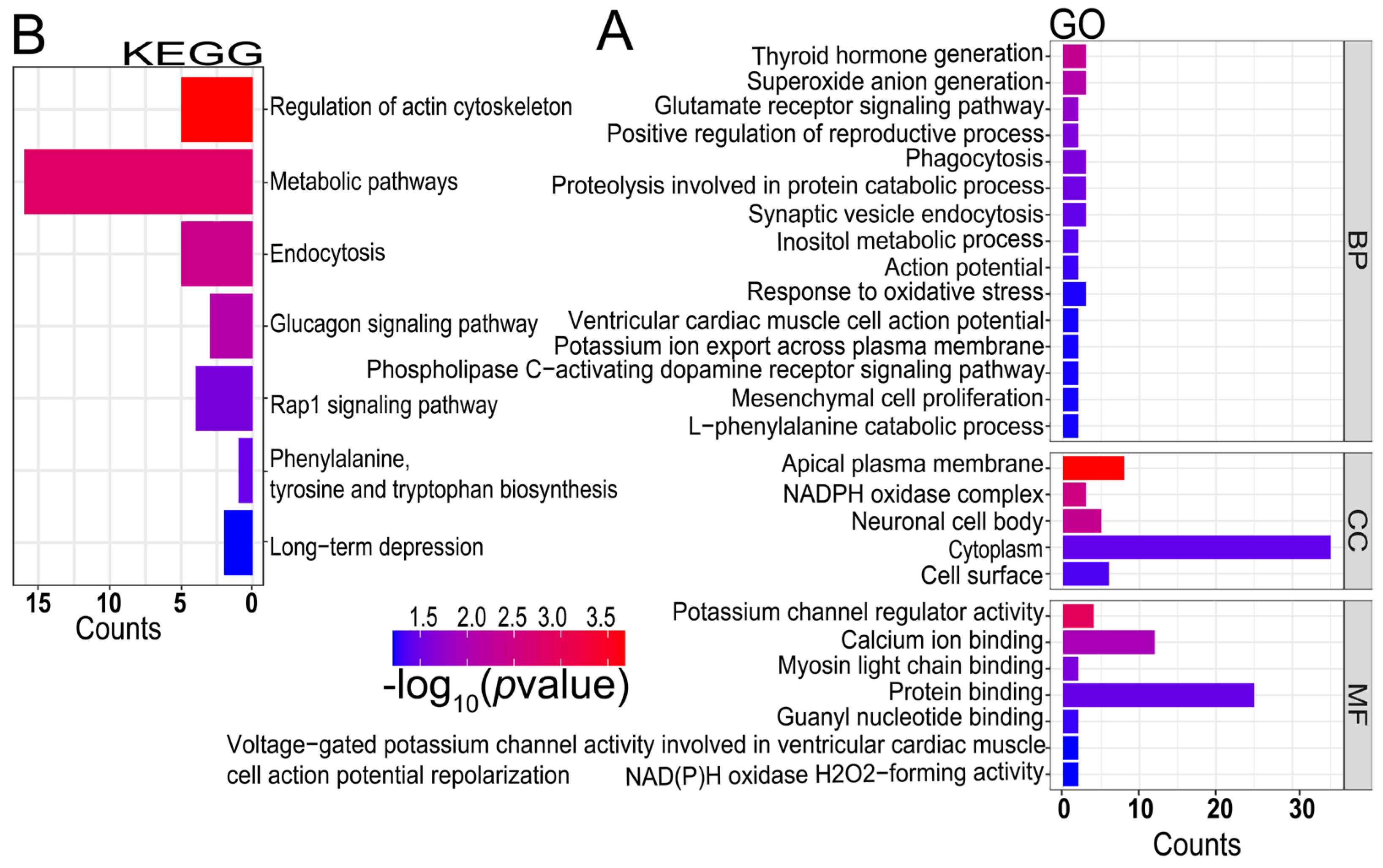
3.4. Candidate Genes Associated with Litter Size Trait
4. Materials and Methods
4.1. Sampling and DNA Extraction
4.2. Genotyping and Quality Control
4.3. Principal Component Analysis and Genetic Diversity
4.4. Runs of Homozygosity Detection
4.5. Inbreeding Coefficients Estimation
4.6. Selection Signatures Identification
4.7. Genome-Wide Association Study
4.8. Gene Annotation and Functional Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amer, P.R.; McEwan, J.C.; Dodds, K.G.; Davis, G.H. Economic values for ewe prolificacy and lamb survival in New Zealand sheep. Livest. Prod. Sci. 1999, 58, 75–90. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Hussain, T.; Dai, C.; Li, J.; Huang, P.; Li, Y.; Ding, X.; Huang, J.; Ji, F.; et al. Effects of dietary energy level on growth performance, blood parameters and meat quality in fattening male Hu lambs. J. Anim. Physiol. Anim. Nutr. 2020, 104, 418–430. [Google Scholar] [CrossRef]
- Wang, W.; Liu, S.; Li, F.; Pan, X.; Li, C.; Zhang, X.; Ma, Y.; La, Y.; Xi, R.; Li, T. Polymorphisms of the Ovine BMPR-IB, BMP-15 and FSHR and Their Associations with Litter Size in Two Chinese Indigenous Sheep Breeds. Int. J. Mol. Sci. 2015, 16, 11385–11397. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Z.; Fang, Y.; Cao, C.; Zhang, Z.; Pan, Y.; Wang, Q. Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep. Genes 2022, 13, 1848. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.P.; Silva, M.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Yin, C.; Wang, Y.; Zhou, P.; Shi, H.; Ma, X.; Yin, Z.; Liu, Y. Genomic Scan for Runs of Homozygosity and Selective Signature Analysis to Identify Candidate Genes in Large White Pigs. Int. J. Mol. Sci. 2023, 24, 12914. [Google Scholar] [CrossRef] [PubMed]
- Rajawat, D.; Nayak, S.S.; Jain, K.; Sharma, A.; Parida, S.; Sahoo, S.P.; Bhushan, B.; Patil, D.B.; Dutt, T.; Panigrahi, M. Genomic patterns of selection in morphometric traits across diverse Indian cattle breeds. Mamm. Genome 2024, 35, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Lukic, B.; Curik, I.; Drzaic, I.; Galić, V.; Shihabi, M.; Vostry, L.; Cubric-Curik, V. Genomic signatures of selection, local adaptation and production type characterisation of East Adriatic sheep breeds. J. Anim. Sci. Biotechnol. 2023, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Ramljak, J.; Špehar, M.; Ceranac, D.; Držaić, V.; Pocrnić, I.; Barać, D.; Mioč, B.; Širić, I.; Barać, Z.; Ivanković, A.; et al. Genomic Characterization of Local Croatian Sheep Breeds-Effective Population Size, Inbreeding & Signatures of Selection. Animals 2024, 14, 1928. [Google Scholar] [CrossRef]
- Wang, R.; Wang, X.; Qi, Y.; Li, Y.; Na, Q.; Yuan, H.; Rong, Y.; Ao, X.; Guo, F.; Zhang, L.; et al. Genetic diversity analysis of Inner Mongolia cashmere goats (Erlangshan subtype) based on whole genome re-sequencing. BMC Genom. 2024, 25, 698. [Google Scholar] [CrossRef]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Saravanan, K.; Panigrahi, M.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B. Selection signatures in livestock genome: A review of concepts, approaches and applications. Livest. Sci. 2020, 241, 104257. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Nayak, S.S.; Panigrahi, M.; Rajawat, D.; Ghildiyal, K.; Sharma, A.; Jain, K.; Bhushan, B.; Dutt, T. Deciphering climate resilience in Indian cattle breeds by selection signature analyses. Trop. Anim. Health Prod. 2024, 56, 46. [Google Scholar] [CrossRef]
- Zhao, F.; Xie, R.; Fang, L.; Xiang, R.; Yuan, Z.; Liu, Y.; Wang, L. Analysis of 206 whole-genome resequencing reveals selection signatures associated with breed-specific traits in Hu sheep. Evol. Appl. 2024, 17, e13697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Zhao, F.; Ren, H.; Xu, L.; Lu, J.; Zhang, S.; Zhang, X.; Wei, C.; Lu, G.; et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS ONE 2013, 8, e66569. [Google Scholar] [CrossRef]
- Zhu, M.; Yang, Y.; Yang, H.; Zhao, Z.; Zhang, H.; Blair, H.T.; Zheng, W.; Wang, M.; Fang, C.; Yu, Q.; et al. Whole-genome resequencing of the native sheep provides insights into the microevolution and identifies genes associated with reproduction traits. BMC Genom. 2023, 24, 392. [Google Scholar] [CrossRef]
- Li, J.; Sun, L.; Sun, J.; Jiang, H. Genome-wide association research on the reproductive traits of Qianhua Mutton Merino sheep. Anim. Biosci. 2024, 37, 1535–1546. [Google Scholar] [CrossRef]
- Smitchger, J.A.; Taylor, J.B.; Mousel, M.R.; Schaub, D.; Thorne, J.W.; Becker, G.M.; Murdoch, B.M. Genome-wide associations with longevity and reproductive traits in U.S. rangeland ewes. Front. Genet. 2024, 15, 1398123. [Google Scholar] [CrossRef] [PubMed]
- Piper, L.R.; Bindon, B.M. The Booroola Merino and the performance of medium non-Peppin crosses at Armidale. Wool Technol. Sheep Breed. 1982, 31, 14. [Google Scholar]
- Davis, G.H. Major genes affecting ovulation rate in sheep. Genet. Sel. Evol. 2005, 37 (Suppl. S1), S11–S23. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.; Zhong, R.; Sun, W.; Chu, M. Expression profiles of oviductal mRNAs and lncRNAs in the follicular phase and luteal phase of sheep (Ovis aries) with 2 fecundity gene (FecB) genotypes. G3: Genes Genomes Genet. 2023, 14, jkad270. [Google Scholar] [CrossRef] [PubMed]
- Bravo, S.; Larama, G.; Paz, E.; Inostroza, K.; Montaldo, H.H.; Sepúlveda, N. Polymorphism of the GDF9 gene associated with litter size in Araucana creole sheep. Anim. Genet. 2016, 47, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Cao, Z.; Hao, Q.; He, M.; Cang, M.; Yu, H.; Ma, Q.; Li, X.; Bao, S.; Wang, J.; et al. Effects of New Mutations in BMPRIB, GDF9, BMP15, LEPR, and B4GALNT2 Genes on Litter Size in Sheep. Vet. Sci. 2023, 10, 258. [Google Scholar] [CrossRef]
- Galloway, S.M.; McNatty, K.P.; Cambridge, L.M.; Laitinen, M.P.; Juengel, J.L.; Jokiranta, T.S.; McLaren, R.J.; Luiro, K.; Dodds, K.G.; Montgomery, G.W.; et al. Mutations in an oocyte-derived growth factor gene (BMP15) cause increased ovulation rate and infertility in a dosage-sensitive manner. Nat. Genet. 2000, 25, 279–283. [Google Scholar] [CrossRef]
- Heaton, M.P.; Leymaster, K.A.; Kalbfleisch, T.S.; Kijas, J.W.; Clarke, S.M.; McEwan, J.; Maddox, J.F.; Basnayake, V.; Petrik, D.T.; Simpson, B.; et al. SNPs for parentage testing and traceability in globally diverse breeds of sheep. PLoS ONE 2014, 9, e94851. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Jensen, J.D. Effects of Selection at Linked Sites on Patterns of Genetic Variability. Annu. Rev. Ecol. Evol. Syst. 2021, 52, 177–197. [Google Scholar] [CrossRef]
- Gravel, S.; Henn, B.M.; Gutenkunst, R.N.; Indap, A.R.; Marth, G.T.; Clark, A.G.; Yu, F.; Gibbs, R.A.; Bustamante, C.D. Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. USA 2011, 108, 11983–11988. [Google Scholar] [CrossRef]
- Liu, J.; Shi, L.; Li, Y.; Chen, L.; Garrick, D.; Wang, L.; Zhao, F. Estimates of genomic inbreeding and identification of candidate regions that differ between Chinese indigenous sheep breeds. J. Anim. Sci. Biotechnol. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Pu, Y.; Liang, B.; Bai, T.; Liu, Y.; Jiang, L.; Ma, Y. A study using single-locus and multi-locus genome-wide association study to identify genes associated with teat number in Hu sheep. Anim. Genet. 2022, 53, 203–211. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef]
- Wright, S. Coefficients of inbreeding and relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef]
- Dadousis, C.; Ablondi, M.; Cipolat-Gotet, C.; van Kaam, J.T.; Finocchiaro, R.; Marusi, M.; Cassandro, M.; Sabbioni, A.; Summer, A. Genomic inbreeding coefficients using imputation genotypes: Assessing the effect of ancestral genotyping in Holstein-Friesian dairy cows. J. Dairy Sci. 2024, 107, 5869–5880. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.; Wu, X.Y.; Juengel, J.L.; Ross, I.K.; Lumsden, J.M.; Lord, E.A.; Dodds, K.G.; Walling, G.A.; McEwan, J.C.; O’Connell, A.R.; et al. Highly prolific Booroola sheep have a mutation in the intracellular kinase domain of bone morphogenetic protein IB receptor (ALK-6) that is expressed in both oocytes and granulosa cells. Biol. Reprod. 2001, 64, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Pan, Z.; Wang, X.; Hu, W.; Di, R.; Yao, Y.; Chu, M. Progress on major genes for high fecundity in ewes. Front. Agric. Sci. Eng. 2014, 1, 9. [Google Scholar] [CrossRef]
- Liu, Z.; Ji, Z.; Wang, G.; Chao, T.; Hou, L.; Wang, J. Genome-wide analysis reveals signatures of selection for important traits in domestic sheep from different ecoregions. BMC Genom. 2016, 17, 863. [Google Scholar] [CrossRef]
- Lv, X.; Chen, W.; Wang, S.; Cao, X.; Yuan, Z.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. Whole-genome resequencing of Dorper and Hu sheep to reveal selection signatures associated with important traits. Anim. Biotechnol. 2023, 34, 3016–3026. [Google Scholar] [CrossRef]
- Abied, A.; Xu, L.; Sahlu, B.W.; Xing, F.; Ahbara, A.; Pu, Y.; Lin, J.; Berihulay, H.; Islam, R.; He, X.; et al. Genome-Wide Analysis Revealed Homozygosity and Demographic History of Five Chinese Sheep Breeds Adapted to Different Environments. Genes 2020, 11, 1480. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Hou, D.; Zhao, Q.; Zhan, S.; Wang, L.; Li, L.; Zhang, H.; Zhao, W.; Yang, S.; Niu, L. Comparative whole-genome resequencing to uncover selection signatures linked to litter size in Hu Sheep and five other breeds. BMC Genom. 2024, 25, 480. [Google Scholar] [CrossRef]
- Mulsant, P.; Lecerf, F.; Fabre, S.; Schibler, L.; Monget, P.; Lanneluc, I.; Pisselet, C.; Riquet, J.; Monniaux, D.; Callebaut, I.; et al. Mutation in bone morphogenetic protein receptor-IB is associated with increased ovulation rate in Booroola Mérino ewes. Proc. Natl. Acad. Sci. USA 2001, 98, 5104–5109. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Gotoh, Y.; Kawahara, T.; Sugimoto, Y. Molecular Effects of Polymorphism in the 3′UTR of Unc-5 homolog C Associated with Conception Rate in Holsteins. PLoS ONE 2015, 10, e0131283. [Google Scholar] [CrossRef] [PubMed]
- Eydivandi, S.; Roudbar, M.A.; Karimi, M.O.; Sahana, G. Genomic scans for selective sweeps through haplotype homozygosity and allelic fixation in 14 indigenous sheep breeds from Middle East and South Asia. Sci. Rep. 2021, 11, 2834. [Google Scholar] [CrossRef]
- Wang, J.; Pan, Y.; Zhang, R.; Xu, G.; Wu, R.; Zhang, W.; Wang, X.; Su, X.; Si, Q.; Yu, S. Expression and Localization of Fas-Associated Factor 1 in Testicular Tissues of Different Ages and Ovaries at Different Reproductive Cycle Phases of Bos grunniens. Animals 2023, 13, 340. [Google Scholar] [CrossRef]
- Cai, X.; Yu, S.; Mipam, T.; Yang, F.; Zhao, W.; Liu, W.; Cao, S.; Shen, L.; Zhao, F.; Sun, L.; et al. Comparative analysis of testis transcriptomes associated with male infertility in cattleyak. Theriogenology 2017, 88, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.T.; Gatti, D.M.; Philip, V.M.; Kasparek, S.; Kreuzman, A.M.; Mansky, B.; Sharif, K.; Taterra, D.; Taylor, W.M.; Thomas, M.; et al. Genome-wide association for testis weight in the diversity outbred mouse population. Mamm. Genome 2018, 29, 310–324. [Google Scholar] [CrossRef]
- Omolaoye, T.S.; Omolaoye, V.A.; Kandasamy, R.K.; Hachim, M.Y.; Du Plessis, S.S. Omics and Male Infertility: Highlighting the Application of Transcriptomic Data. Life 2022, 12, 280. [Google Scholar] [CrossRef]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimarães, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50, 40. [Google Scholar] [CrossRef]
- Bansal, S.K.; Gupta, N.; Sankhwar, S.N.; Rajender, S. Differential Genes Expression between Fertile and Infertile Spermatozoa Revealed by Transcriptome Analysis. PLoS ONE 2015, 10, e0127007. [Google Scholar] [CrossRef]
- Hodge, M.J.; de Las Heras-Saldana, S.; Rindfleish, S.J.; Stephen, C.P.; Pant, S.D. QTLs and Candidate Genes Associated with Semen Traits in Merino Sheep. Animals 2023, 13, 2286. [Google Scholar] [CrossRef]
- Yang, R.; Duan, C.; Zhang, S.; Liu, Y.; Zhang, Y. Prolactin Regulates Ovine Ovarian Granulosa Cell Apoptosis by Affecting the Expression of MAPK12 Gene. Int. J. Mol. Sci. 2023, 24, 10269. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, S.; Boruah, P.; Xie, L.; Kulyar, M.F.; Nawaz, M.; Yousuf, S.; Liu, T.; Jabeen, F.; Miao, X. Genome-wide transcriptome profiling uncovers differential miRNAs and lncRNAs in ovaries of Hu sheep at different developmental stages. Sci. Rep. 2021, 11, 5865. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, H.; Yang, H.; Zhao, Z.; Blair, H.T.; Liang, H.; Wu, P.; Yu, Q. Targeting GNAQ in hypothalamic nerve cells to regulate seasonal estrus in sheep. Theriogenology 2022, 181, 79–88. [Google Scholar] [CrossRef]
- Tang, E.I.; Xiao, X.; Mruk, D.D.; Qian, X.J.; Mok, K.W.; Jenardhanan, P.; Lee, W.M.; Mathur, P.P.; Cheng, C.Y. Microtubule affinity-regulating kinase 4 (MARK4) is a component of the ectoplasmic specialization in the rat testis. Spermatogenesis 2012, 2, 117–126. [Google Scholar] [CrossRef]
- Tang, E.I.; Cheng, C.Y. MARK2 and MARK4 Regulate Sertoli Cell BTB Dynamics through Microtubule and Actin Cytoskeletons. Endocrinology 2022, 163, bqac130. [Google Scholar] [CrossRef] [PubMed]
- Fazilat, A.; Rashid, N.; Nigam, A.; Anjum, S.; Gupta, N.; Wajid, S. Differential Expression of MARK4 Protein and Related Perturbations in Females with Ovulatory PCOS. Endocr. Metab. Immune Disord Drug Targets 2019, 19, 1064–1074. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- Pavlidis, P.; Živkovic, D.; Stamatakis, A.; Alachiotis, N. SweeD: Likelihood-based detection of selective sweeps in thousands of genomes. Mol. Biol. Evol. 2013, 30, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Klassmann, A.; Vitalis, R. rehh 2.0: A reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol. Ecol. Resour. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- Gautier, M.; Naves, M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 2011, 20, 3128–3143. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Wang, D.; Kang, H.; Mrode, R.; Zhou, L.; Xu, S.; Liu, J.F. A rapid epistatic mixed-model association analysis by linear retransformations of genomic estimated values. Bioinformatics 2018, 34, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, J.; Xiong, J.; Huang, J.; Yang, P.; Shang, M.; Zhang, L. Genetic Diversity, Selection Signatures, and Genome-Wide Association Study Identify Candidate Genes Related to Litter Size in Hu Sheep. Int. J. Mol. Sci. 2024, 25, 9397. https://doi.org/10.3390/ijms25179397
Bao J, Xiong J, Huang J, Yang P, Shang M, Zhang L. Genetic Diversity, Selection Signatures, and Genome-Wide Association Study Identify Candidate Genes Related to Litter Size in Hu Sheep. International Journal of Molecular Sciences. 2024; 25(17):9397. https://doi.org/10.3390/ijms25179397
Chicago/Turabian StyleBao, Jingjing, Jinke Xiong, Jupeng Huang, Peifu Yang, Mingyu Shang, and Li Zhang. 2024. "Genetic Diversity, Selection Signatures, and Genome-Wide Association Study Identify Candidate Genes Related to Litter Size in Hu Sheep" International Journal of Molecular Sciences 25, no. 17: 9397. https://doi.org/10.3390/ijms25179397