

Genome-Wide Association Study of Resistance to Largemouth Bass Ranavirus (LMBV) in Micropterus salmoides

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Experimental Challenge and Samples
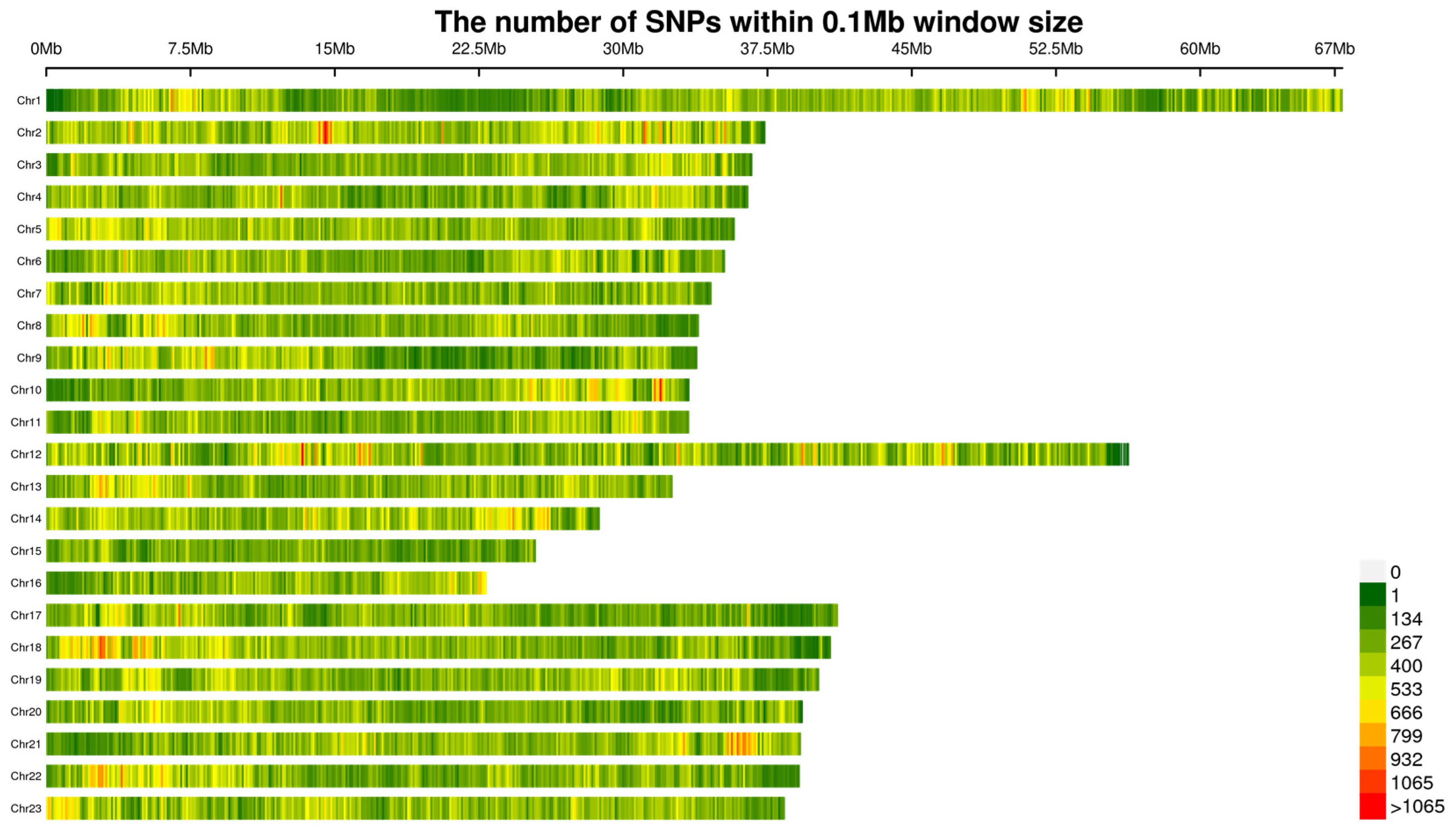
2.2. SNP Data and Density on Chromosomes
2.3. Analysis of the Linkage Disequilibrium Population Structure
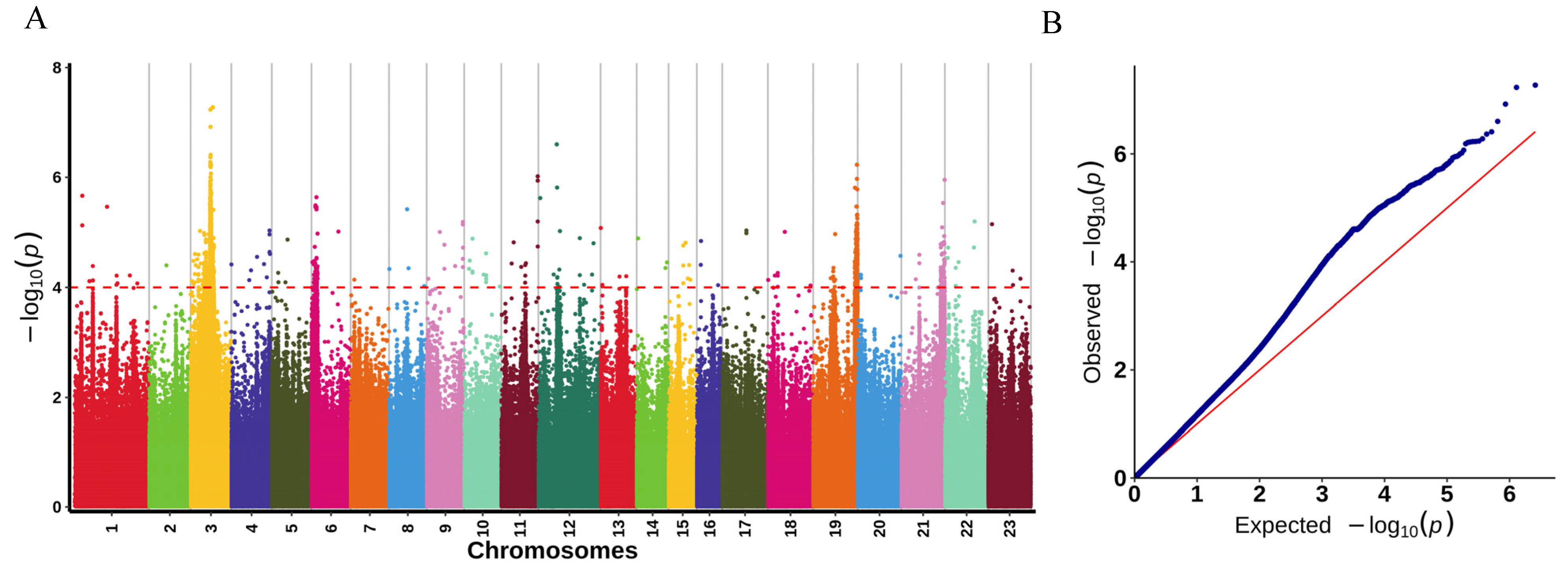
2.4. GWAS for LMBV Resistance
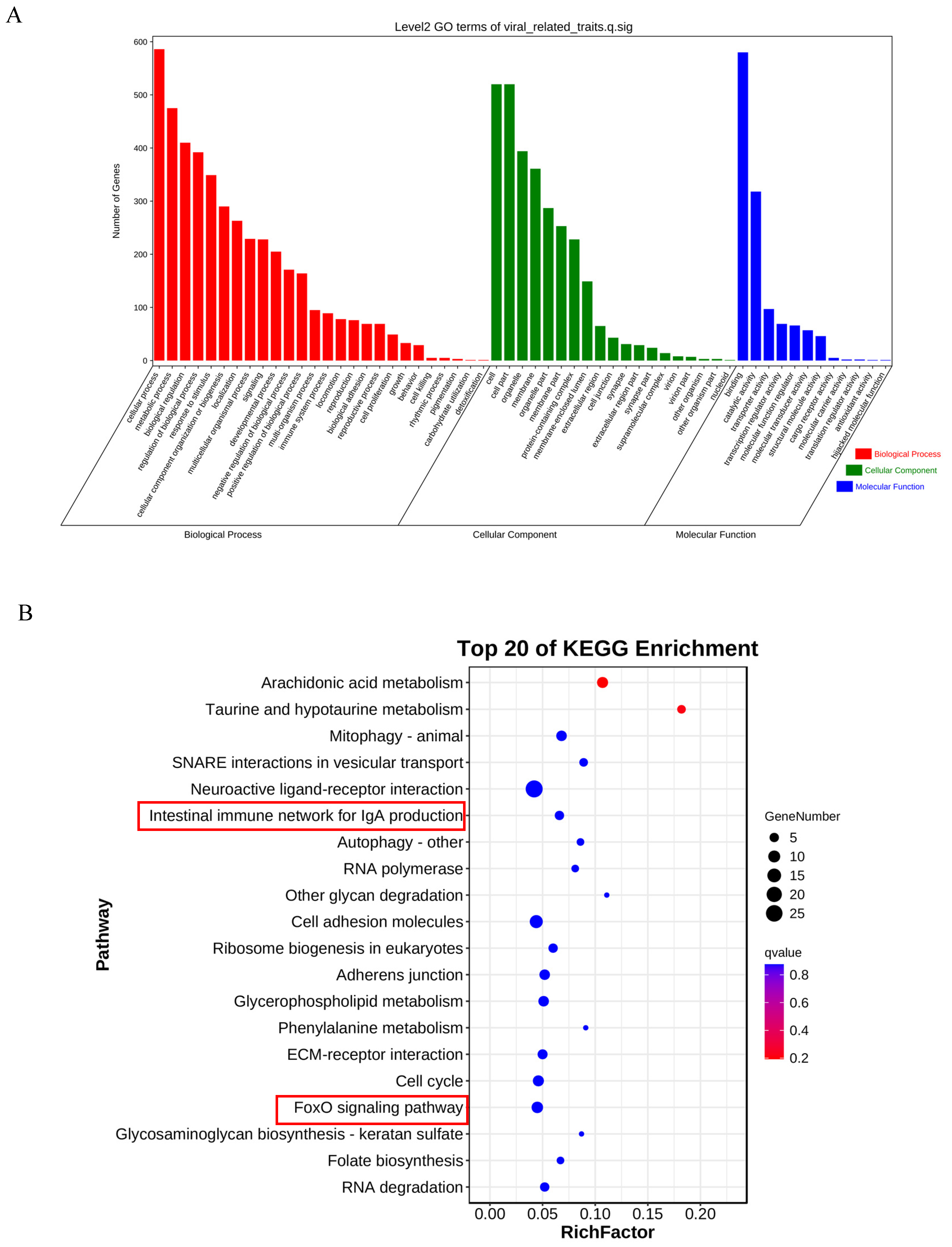
2.5. GO/KEGG Enrichment of Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Fish and LMBV Challenge
4.2. Variant Identification and Annotation
4.3. Linkage Disequilibrium and Population Structure Analysis
4.4. Genome-Wide Association Study (GWAS)
4.5. Annotation of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAO. The State of world fisheries and aquaculture 2024. In Blue Transformation in Action; FAO: Rome, Italy, 2024. [Google Scholar] [CrossRef]
- Dong, C.; Wang, Z.; Weng, S.; He, J. Occurrence of a lethal ranavirus in hybrid mandarin (Siniperca scherzeri × Siniperca chuatsi) in Guangdong, South China. Vet. Microbiol. 2017, 203, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Geng, Y.; Qin, Z.; Wang, K.; Ouyang, P.; Chen, D.; Huang, X.; Zuo, Z.; He, C.; Guo, H.; et al. A new ranavirus of the santee-cooper group invades largemouth bass (Micropterus salmoides) culture in southwest China. Aquaculture 2020, 526, 735363. [Google Scholar] [CrossRef]
- Grant, E.C.; Philipp, D.P.; Inendino, K.R.; Goldberg, T.L. Effects of temperature on the susceptibility of largemouth bass to largemouth bass virus. J. Aquat. Anim. Health 2003, 15, 215–220. [Google Scholar] [CrossRef]
- Inendino, K.R.; Grant, E.C.; Philipp, D.P.; Goldberg, T.L. Effects of factors related to water quality and population density on the sensitivity of juvenile largemouth bass to mortality induced by viral infection. J. Aquat. Anim. Health 2005, 17, 304–314. [Google Scholar] [CrossRef]
- Getchell, R.G.; Groocock, G.H.; Schumacher, V.L.; Grimmett, S.G.; Wooster, G.A.; Bowser, P.R. Quantitative polymerase chain reaction assay for largemouth bass virus. J. Aquat. Anim. Health 2007, 19, 226–233. [Google Scholar] [CrossRef]
- Grizzle, J.; Altinok, I.; Noyes, A. PCR method for detection of largemouth bass virus. Dis. Aquat. Organ. 2003, 54, 29–33. [Google Scholar] [CrossRef]
- Jin, R.; Zhai, L.; Zhu, Q.; Feng, J.; Pan, X. Naked-eyes detection of largemouth bass ranavirus in clinical fish samples using gold nanoparticles as colorimetric sensor. Aquaculture 2020, 528, 735554. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Li, J.; Huang, X.; Wei, J.; Yang, J.; Guan, L.; Wen, X.; Wang, S.; Qin, Q. A novel sandwich elasa based on aptamer for detection of largemouth bass virus (LMBV). Viruses 2022, 14, 945. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, Y.; Feng, J. Rapid diagnosis of largemouth bass ranavirus in fish samples using the loop-mediated isothermal amplification method. Mol. Cell. Probes. 2020, 52, 101569. [Google Scholar] [CrossRef]
- Liu, X.; He, M.-S.; Yang, K.-C.; Yang, B.; Ling, F.; Wang, G.-X. Mesoporous Silica nanocarriers loaded with ribavirin against largemouth bass virus. Aquaculture 2023, 564, 739078. [Google Scholar] [CrossRef]
- Yi, W.; Zhang, X.; Zeng, K.; Xie, D.; Song, C.; Tam, K.; Liu, Z.; Zhou, T.; Li, W. Construction of a DNA vaccine and its protective effect on largemouth bass (Micropterus salmoides) challenged with largemouth bass virus (LMBV). Fish Shellfish Immunol. 2020, 106, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Fu, X.; Lin, Q.; Liang, H.; Luo, X.; Zuo, S.; Liu, L.; Li, N. In vivo and in vitro, antiviral effects of two mixture of Chinese herbal drug active monomers against MSRV and LMBV in largemouth bass (Micropterus salmoides). Aquaculture 2023, 577, 739977. [Google Scholar] [CrossRef]
- Yue, G.H. Recent Advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish. 2014, 15, 376–396. [Google Scholar] [CrossRef]
- Zenger, K.R.; Khatkar, M.S.; Jones, D.B.; Khalilisamani, N.; Jerry, D.R.; Raadsma, H.W. Genomic selection in aquaculture: Application, limitations and opportunities with special reference to marine shrimp and pearl oysters. Front. Genet. 2019, 9, 693. [Google Scholar] [CrossRef]
- Rasal, K.D.; Chakrapani, V.; Pandey, A.K.; Rasal, A.R.; Sundaray, J.K.; Ninawe, A.; Jayasankar, P. Status and future perspectives of single nucleotide polymorphisms (SNPs) markers in farmed fishes: Way ahead using next generation sequencing. Gene Rep. 2017, 6, 81–86. [Google Scholar] [CrossRef]
- Wenne, R. Single nucleotide polymorphism markers with applications in aquaculture and assessment of its impact on natural populations. Aquat. Living Resour. 2018, 31, 2. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A Review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, J.; Zou, Z.; Long, C.; Lin, J.; Zeng, J.; Hou, J.; Han, L.; Jiang, Y.; Li, S.; et al. Identification of SNPs and candidate genes associate with growth performance in all-female mandarin fish (Siniperca chuatsi) by a genome-wide association study. Aquaculture 2024, 586, 740778. [Google Scholar] [CrossRef]
- Cáceres, P.; Lopéz, P.; Garcia, B.; Cichero, D.; Ødegård, J.; Moen, T.; Yáñez, J.M. Meta-analysis of GWAS for sea lice load in Atlantic Salmon. Aquaculture 2024, 584, 740543. [Google Scholar] [CrossRef]
- Rodríguez, F.H.; Flores-Mara, R.; Yoshida, G.M.; Barría, A.; Jedlicki, A.M.; Lhorente, J.P.; Reyes-López, F.; Yáñez, J.M. Genome-wide association analysis for resistance to infectious pancreatic necrosis virus identifies candidate genes involved in viral replication and immune response in rainbow trout (Oncorhynchus Mykiss). G3-Genes Genomes Genet. 2019, 9, 2897–2904. [Google Scholar] [CrossRef]
- Vallejo, R.L.; Cheng, H.; Fragomeni, B.O.; Shewbridge, K.L.; Gao, G.; MacMillan, J.R.; Towner, R.; Palti, Y. Genome-wide association analysis and accuracy of genome-enabled breeding value predictions for resistance to infectious hematopoietic necrosis virus in a commercial rainbow trout breeding population. Genet. Sel. Evol. 2019, 51, 47. [Google Scholar] [CrossRef] [PubMed]
- Palaiokostas, C.; Cariou, S.; Bestin, A.; Bruant, J.-S.; Haffray, P.; Morin, T.; Cabon, J.; Allal, F.; Vandeputte, M.; Houston, R.D. Genome-wide association and genomic prediction of resistance to viral nervous necrosis in European sea bass (Dicentrarchus labrax) using RAD sequencing. Genet. Sel. Evol. 2018, 50, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, P.; Huang, S.; Ye, B.; Chua, E.; Wan, Z.Y.; Yue, G.H. Genome-wide association study identifies loci associated with resistance to viral nervous necrosis disease in Asian seabass. Mar. Biotechnol. 2017, 19, 255–265. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, T.; Bai, H.; Ke, Q.; Li, B.; Bai, M.; Zhou, Z.; Pu, F.; Zheng, W.; Xu, P. Genome-wide association analysis reveals the genetic architecture of parasite (Cryptocaryon irritans) resistance in large yellow croaker (Larimichthys crocea). Mar. Biotechnol. 2021, 23, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Sha, J.; Liu, S.; Bao, L.; Zhang, J.; Wang, R.; Yao, J.; Li, C.; Feng, J.; Sun, F.; et al. A genome-wide association study in catfish reveals the presence of functional hubs of related genes within QTLs for columnaris disease resistance. BMC Genom. 2015, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Su, Z.; Li, Y.; Liu, Y.; Wang, L.; Lu, S.; Wang, S.; Gan, T.; Liu, F.; Zhou, X.; et al. Genome-wide association mapping and gene expression analyses reveal genetic mechanisms of disease resistance variations in Cynoglossus semilaevis. Front. Genet. 2019, 10, 1167. [Google Scholar] [CrossRef] [PubMed]
- Lira, L.V.G.; Mastrochirico-Filho, V.A.; Mendes, N.J.; Ariede, R.B.; Yáñez, J.M.; Hashimoto, D.T. Genome-wide association study of host resistance to the ectoparasite ichthyophthirius multifiliis in the Amazon fish Colossoma macropomum. Mol. Biol. Rep. 2023, 50, 599–607. [Google Scholar] [CrossRef]
- Barría, A.; Trịnh, T.Q.; Mahmuddin, M.; Peñaloza, C.; Papadopoulou, A.; Gervais, O.; Chadag, V.M.; Benzie, J.A.H.; Houston, R.D. A major quantitative trait locus affecting resistance to tilapia lake virus in farmed nile tilapia (Oreochromis niloticus). Heredity 2021, 127, 334–343. [Google Scholar] [CrossRef]
- Liyanage, D.S.; Lee, S.; Yang, H.; Lim, C.; Omeka, W.K.M.; Sandamalika, W.M.G.; Udayantha, H.M.V.; Kim, G.; Ganeshalingam, S.; Jeong, T.; et al. Genome-wide association study of VHSV-resistance trait in Paralichthys olivaceus. Fish Shellfish Immunol. 2022, 124, 391–400. [Google Scholar] [CrossRef]
- Fuji, K.; Hasegawa, O.; Honda, K.; Kumasaka, K.; Sakamoto, T.; Okamoto, N. Marker-assisted breeding of a lymphocystis disease-resistant Japanese flounder (Paralichthys olivaceus). Aquaculture 2007, 272, 291–295. [Google Scholar] [CrossRef]
- Sawayama, E.; Kitamura, S.-I.; Nakayama, K.; Ohta, K.; Okamoto, H.; Ozaki, A.; Takagi, M. Development of a novel RSIVD-resistant strain of red sea bream (Pagrus major) by marker-assisted selection combined with DNA-based family selection. Aquaculture 2019, 506, 188–192. [Google Scholar] [CrossRef]
- Sun, C.; Li, J.; Dong, J.; Niu, Y.; Hu, J.; Lian, J.; Li, W.; Li, J.; Tian, Y.; Shi, Q.; et al. Chromosome-level genome assembly for the largemouth bass Micropterus salmoides provides insights into adaptation to fresh and brackish water. Mol. Ecol. Resour. 2021, 21, 301–315. [Google Scholar] [CrossRef] [PubMed]
- San, L.; Liu, B.; Liu, B.; Zhu, K.; Guo, L.; Guo, H.; Zhang, N.; Jiang, S.; Zhang, D. Genome-wide association study reveals multiple novel SNPs and putative candidate genes associated with low oxygen tolerance in golden pompano Trachinotus ovatus (Linnaeus 1758). Aquaculture 2021, 544, 737098. [Google Scholar] [CrossRef]
- Tang, H.; Liu, J.; Wang, Z.; Zhang, L.; Yang, M.; Huang, J.; Wen, X.; Luo, J. Genome-wide association study (GWAS) analysis of black color trait in the leopard coral grouper (Plectropomus leopardus) using whole genome resequencin. Comp. Biochem. Physiol. Part D Genom. Proteom. 2023, 48, 101138. [Google Scholar] [CrossRef] [PubMed]
- Bishop, S.C.; Woolliams, J.A. Genomics and disease resistance studies in livestock. Livest. Sci. 2014, 166, 190–198. [Google Scholar] [CrossRef]
- Duan, X.; Liang, K.; Yang, M.; Zhang, M.; Zuo, X.; Jia, X.; Li, Z.; Yu, J.; Luo, L.; Shan, J.; et al. Genome-wide association study identifies candidate SNPs and genes associated with red-spotted grouper nervous necrosis virus infection of the giant grouper (Epinephelus lanceolatus). Aquaculture 2024, 578, 740126. [Google Scholar] [CrossRef]
- Jin, R.; Huang, H.; Zhou, Y.; Wang, Y.; Fu, H.; Li, Z.; Fu, X.; Li, N. Characterization of mandarin fish (Siniperca chuatsi) IL-6 and IL-6 signal transducer and the association between their SNPs and resistance to ISKNV disease. Fish Shellfish Immunol. 2021, 113, 139–147. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a system understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Sawayama, E.; Tanizawa, S.; Kitamura, S.-I.; Nakayama, K.; Ohta, K.; Ozaki, A.; Takagi, M. Identification of quantitative trait loci for resistance to RSIVD in red sea bream (Pagrus major). Mar. Biotechnol. 2017, 19, 601–613. [Google Scholar] [CrossRef]
- Hillestad, B.; Moghadam, H.K. Genome-wide association study of piscine myocarditis virus (PMCV) resistance in Atlantic salmon (Salmo salar). J. Hered. 2019, 110, 720–726. [Google Scholar] [CrossRef]
- Xu, T.; Chen, S.; Zhang, Y. MHC class IIα gene polymorphism and its association with resistance/susceptibility to Vibrio anguillarum in Japanese flounder (Paralichthys olivaceus). Dev. Comp. Immunol. 2010, 34, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zou, F.; Xin, G.; Xiang, B.-L.; Zhao, J.-Q.; Yuan, S.-F.; Zhang, X.-L.; Zhang, Z.-H. STS IIA inhibited angiogenesis of lung adenocarcinoma by activating FOXO3 to inhibit CXCL1/STAT3/VEGF pathway. Toxicon 2024, 240, 107627. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.R.; Ali, F.E.M.; Abd-Elhamid, T.H.; Hassanein, E.H.M. Coenzyme Q10 protects hepatocytes from ischemia reperfusion-induced apoptosis and oxidative stress via regulation of Bax/Bcl-2/PUMA and Nrf-2/FOXO-3/Sirt-1 signaling pathways. Tissue Cell 2019, 60, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Peng, K.; He, Y.; Xue, L. Mechanistic regulation of FOXO transcription factors in the nucleus. Biochim. Biophys. Acta BBA-Rev. Cancer 2024, 1879, 189083. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, F.; Wang, L.; Gao, M.; Xie, Y.; Sun, Y.; Liu, H.; Yuan, Y.; Yi, W.; Huang, Z.; et al. Virus-induced p38 mapk activation facilitates viral infection. Theranostics 2020, 10, 12223–12240. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Q.; Liu, A.; Zhang, C.; Liu, L.-H.; Lu, L.-F.; Tu, J.; Zhang, Y.-A. MicroRNA miR-155 inhibits cyprinid herpesvirus 3 replication via regulating AMPK-MAVS-IFN axis. Dev. Comp. Immunol. 2022, 129, 104335. [Google Scholar] [CrossRef]
- Kook, I.; Jones, C. The serum and glucocorticoid-regulated protein kinases (SGK) stimulate bovine herpesvirus 1 and herpes simplex virus 1 productive infection. Virus Res. 2016, 222, 106–112. [Google Scholar] [CrossRef]
- Dai, X.; Quan, D.; Wang, L.; Cui, D.; Wan, X.; Ren, Q. FOXO is involved in antimicrobial peptides expression during wssv infection in Exopalaemon carinicauda. Fish Shellfish Immunol. 2024, 144, 109286. [Google Scholar] [CrossRef]
- Li, Y.; Xie, P.; Sun, M.; Xiang, B.; Kang, Y.; Gao, P.; Zhu, W.; Ning, Z.; Ren, T. S1PR1 expression correlates with inflammatory responses to newcastle disease virus infection. Infect. Genet. Evol. 2016, 37, 37–42. [Google Scholar] [CrossRef]
- Corfe, S.A.; Paige, C.J. The many roles of IL-7 in B cell development; mediator of survival, proliferation and differentiation. Semin. Immunol. 2012, 24, 198–208. [Google Scholar] [CrossRef]
- Jiang, C.; Huang, Z.; Tang, L.; Peng, F.; Xiao, Y. Identification and analysis of senescence-related genes in caudal fin cells of triploid crucian carp. Reprod. Breed. 2023, 3, 169–175. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, C.; Wang, B.; Guan, X.; Fang, L.; Zhan, F.; Sun, H.; Li, H.; Lou, C.; Yan, F.; et al. HOXB9 blocks cell cycle progression to inhibit pancreatic cancer cell proliferation through the DNMT1/RBL2/c-Myc axis. Cancer Lett. 2022, 533, 215595. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kohama, Y.; Kuge, A.; Kido, E.; Sakurai, H. GADD45 family proteins suppress JNK signaling by targeting MKK7. Arch. Biochem. Biophys. 2017, 635, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Song, L.; Huang, C. Gadd45 proteins as critical signal transducers linking NF-κB to MAPK cascades. Curr. Cancer Drug Targets 2009, 9, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Liu, R.; Zhang, H.; Zhang, S.; Hu, X.; Tan, J.; Liang, C.; Qiao, W. GADD45 proteins inhibit HIV-1 replication through specific suppression of HIV-1 transcription. Virology 2016, 493, 1–11. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, s13742-015-0047-8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variation | Chromosome | Positon | −log10 (p) | Beta | PVE * |
|---|---|---|---|---|---|
| SNP | Chr3 | 20,046,158 | 7.273655921 | −0.526368 | 0.478266619 |
| Deletion | Chr3 | 17,892,358 | 7.231973449 | −0.6084425 | 0.266545639 |
| SNP | Chr3 | 17,962,533 | 6.922432256 | −0.5461026 | 0.251943056 |
| SNP | Chr12 | 16,554,664 | 6.602898119 | 0.3744209 | 0.277689232 |
| SNP | Chr3 | 18,064,341 | 6.409210533 | −0.5518117 | 0.228920207 |
| SNP | Chr3 | 17,856,093 | 6.366583057 | −0.4706976 | 0.21973947 |
| SNP | Chr3 | 17,752,374 | 6.27981009 | −0.503464 | 0.229345084 |
| SNP | Chr3 | 18,074,846 | 6.238069156 | −0.4931315 | 0.234229701 |
| SNP | Chr19 | 39,488,902 | 6.230333979 | −0.3551792 | 0.224828565 |
| SNP | Chr3 | 18,327,881 | 6.227981378 | −0.5180217 | 0.234803171 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Luo, X.; Zuo, S.; Fu, X.; Lin, Q.; Niu, Y.; Liang, H.; Ma, B.; Li, N. Genome-Wide Association Study of Resistance to Largemouth Bass Ranavirus (LMBV) in Micropterus salmoides. Int. J. Mol. Sci. 2024, 25, 10036. https://doi.org/10.3390/ijms251810036
Li P, Luo X, Zuo S, Fu X, Lin Q, Niu Y, Liang H, Ma B, Li N. Genome-Wide Association Study of Resistance to Largemouth Bass Ranavirus (LMBV) in Micropterus salmoides. International Journal of Molecular Sciences. 2024; 25(18):10036. https://doi.org/10.3390/ijms251810036
Chicago/Turabian StyleLi, Pinhong, Xia Luo, Shaozhi Zuo, Xiaozhe Fu, Qiang Lin, Yinjie Niu, Hongru Liang, Baofu Ma, and Ningqiu Li. 2024. "Genome-Wide Association Study of Resistance to Largemouth Bass Ranavirus (LMBV) in Micropterus salmoides" International Journal of Molecular Sciences 25, no. 18: 10036. https://doi.org/10.3390/ijms251810036