

An 8-SNP LDL Cholesterol Polygenic Score: Associations with Cardiovascular Risk Traits, Familial Hypercholesterolemia Phenotype, and Premature Coronary Heart Disease in Central Romania
 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Genetic Screening for FH Mutations
2.2. Demographic, Clinical, and Biological Characteristics
2.3. Genetic Analysis and Risk Scores
2.4. Establishing the Superior PRS
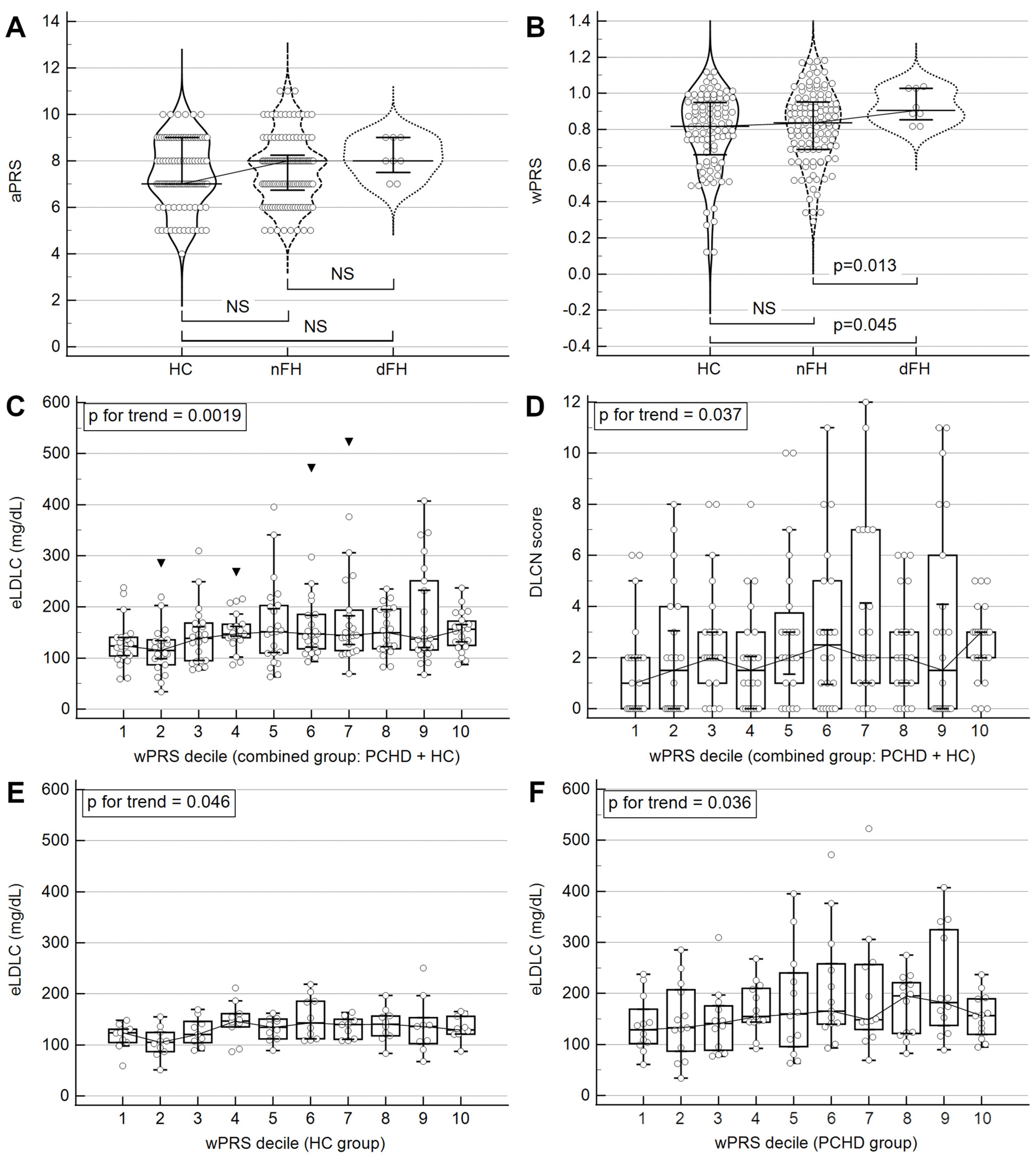
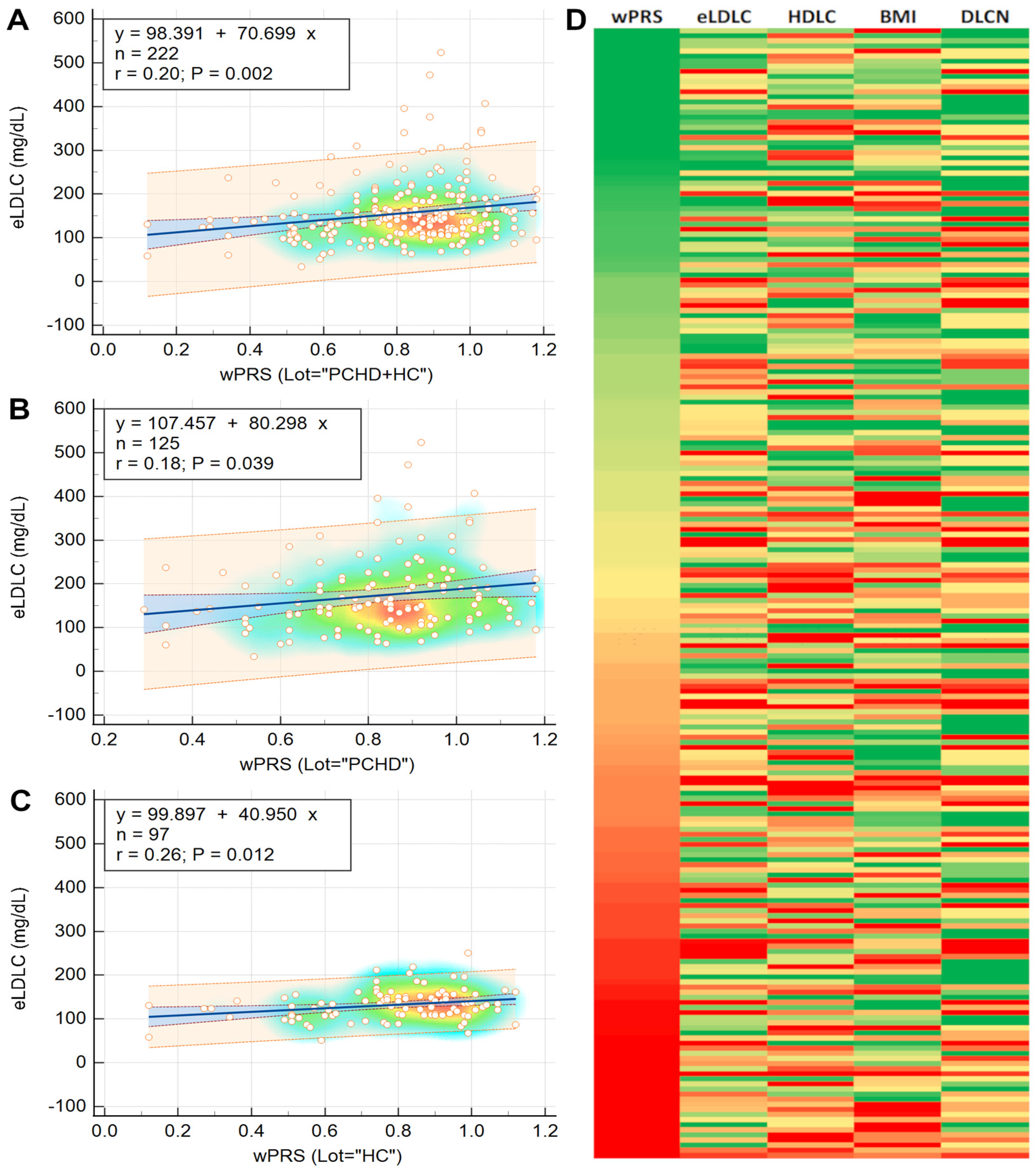
2.5. Phenotypic Traits Associated with wPRS
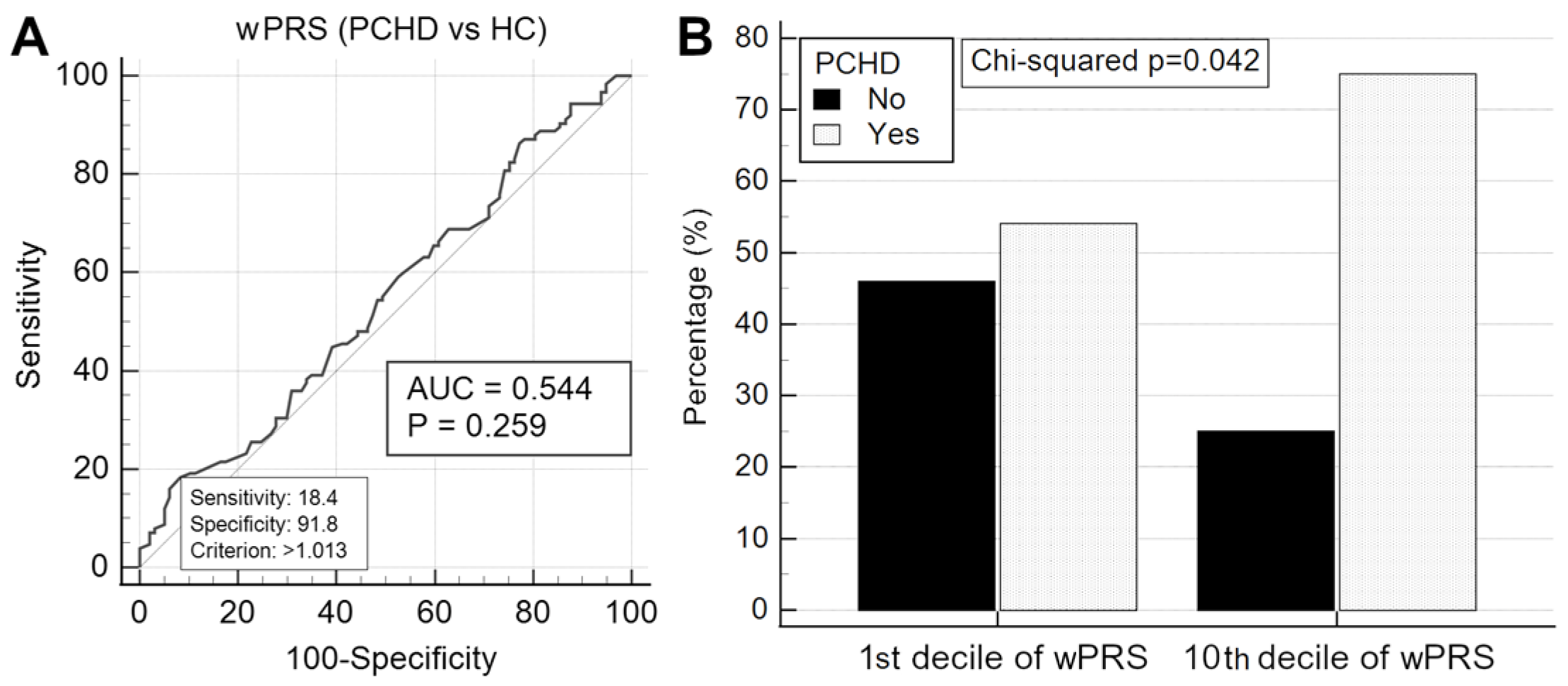
2.6. wPRS Association with PCHD
2.7. Association of wPRS with Clinical FH
3. Discussion
3.1. Monogenic FH
3.2. Genotype–Phenotype Correlation
3.3. Polygenic FH
3.4. LDLC Polygenic Score Is Associated with Phenotypic Traits
3.5. Elevated LDLC Polygenic Score Is Associated with Premature Coronary Heart Disease
3.6. LDLC Polygenic Score Is Associated with Clinical FH Phenotype
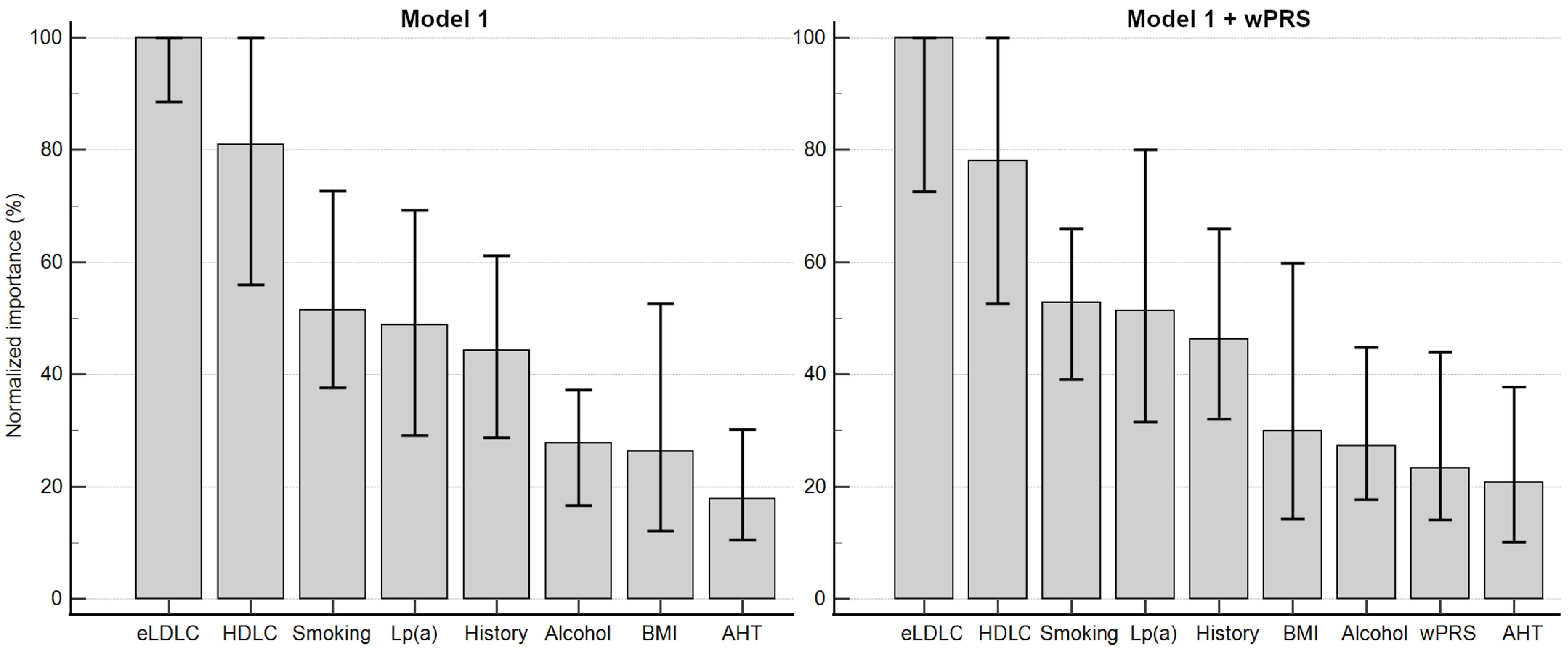
3.7. Clinical Relevance and Implementation of LDLC Polygenic Score
3.8. Strengths and Limitations
4. Materials and Methods
4.1. Study Design and Selection of Participants
4.2. Data Collection from Participants
4.3. Extended Lipid Profile Analysis
4.4. Genetic Analysis and Risk Scores
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khera, A.V.; Kathiresan, S. Genetics of coronary artery disease: Discovery, biology and clinical translation. Nat. Rev. Genet. 2017, 18, 331–344. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.J.; Nabel, E.G. Genomics of Cardiovascular Disease. N. Engl. J. Med. 2011, 365, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Zarkasi, K.A.; Abdullah, N.; Murad, N.A.A.; Ahmad, N.; Jamal, R. Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies. Diagnostics 2022, 12, 2561. [Google Scholar] [CrossRef] [PubMed]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Lao, O.; Lu, T.T.; Nothnagel, M.; Junge, O.; Freitag-Wolf, S.; Caliebe, A.; Balascakova, M.; Bertranpetit, J.; Bindoff, L.A.; Comas, D.; et al. Correlation between Genetic and Geographic Structure in Europe. Curr. Biol. 2008, 18, 1241–1248. [Google Scholar] [CrossRef]
- Moskvina, V.; Smith, M.; Ivanov, D.; Blackwood, D.; StClair, D.; Hultman, C.; Toncheva, D.; Gill, M.; Corvin, A.; O’dushlaine, C.; et al. Genetic Differences between Five European Populations. Hum. Hered. 2010, 70, 141–149. [Google Scholar] [CrossRef]
- Dron, J.S. The clinical utility of polygenic risk scores for combined hyperlipidemia. Curr. Opin. Infect. Dis. 2022, 34, 44–51. [Google Scholar] [CrossRef]
- Trinder, M.; Brunham, L.R. Polygenic scores for dyslipidemia: The emerging genomic model of plasma lipoprotein trait inheritance. Curr. Opin. Infect. Dis. 2020, 32, 103–111. [Google Scholar] [CrossRef]
- Christoffersen, M.; Tybjærg-Hansen, A. Polygenic risk scores: How much do they add? Curr. Opin. Infect. Dis. 2021, 32, 157–162. [Google Scholar] [CrossRef]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef]
- Gratton, J.; Finan, C.; Hingorani, A.D.; Humphries, S.E.; Futema, M. LDL-C Concentrations and the 12-SNP LDL-C Score for Polygenic Hypercholesterolaemia in Self-Reported South Asian, Black and Caribbean Participants of the UK Biobank. Front. Genet. 2022, 13, 845498. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; A Whittall, R.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of Variant Selection for the LDL Cholesterol Genetic Risk Score in the Diagnosis of the Polygenic Form of Clinical Familial Hypercholesterolemia and Replication in Samples from 6 Countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Bourbon, M.; Williams, M.; Humphries, S.E. Clinical utility of the polygenic LDL-C SNP score in familial hypercholesterolemia. Atherosclerosis 2018, 277, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Dobreanu, M.; Moldovan, G.M.; Hadadi, L.; Iancu, M.; Hutanu, A.; Demian, L.; Banescu, C. Preliminary spectrum of familial hypercholes-terolemia gene variants in Romania. Atherosclerosis 2020, 315, E188. [Google Scholar] [CrossRef]
- Moldovan, V.; Bănescu, C.; Dobreanu, M. Genetic Diagnostic Approaches in Familial Hypercholesterolemia Evaluation. Rom. Rev. Lab. Med. 2021, 29, 319–325. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1046–e1081. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of Apolipoprotein E Genotypes with Lipid Levels and Coronary Risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; De Marco, M.; Stevens, C.A.; Akram, A.; Freiberger, T.; Hovingh, G.K.; Kastelein, J.J.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries—The EAS familial hypercholesterolaemia studies collaboration (FHSC). Atherosclerosis 2018, 277, 234–255. [Google Scholar] [CrossRef]
- Reeskamp, L.F.; Tromp, T.R.; Defesche, J.C.; Grefhorst, A.; Stroes, E.S.; Hovingh, G.K.; Zuurbier, L. Next-Generation sequencing to confirm clinical familial hypercholesterolemia. Eur. J. Prev. Cardiol. 2021, 28, 875–883. [Google Scholar] [CrossRef]
- Garg, A.; Fazio, S.; Duell, P.B.; Baass, A.; Udata, C.; Joh, T.; Riel, T.; Sirota, M.; Dettling, D.; Liang, H.; et al. Molecular Characterization of Familial Hypercholesterolemia in a North American Cohort. J. Endocr. Soc. 2020, 4, bvz015. [Google Scholar] [CrossRef]
- Humphries, S.E.; Whittall, R.A.; Hubbart, C.S.; Maplebeck, S.; Cooper, J.A.; Soutar, A.K.; Naoumova, R.; Thompson, G.R.; Seed, M.; Durrington, P.N.; et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: Relation to plasma lipid levels and coronary heart disease risk. J. Med Genet. 2006, 43, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Wang, D.; Patel, K.; Whittall, R.; Wood, G.; Farrer, M.; Neely, R.; Fairgrieve, S.; Nair, D.; Barbir, M.; et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clin. Genet. 2010, 77, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, S.W.; Kastelein, J.J.; Defesche, J.C. Update of the molecular basis of familial hypercholesterolemia in The Netherlands. Hum. Mutat. 2005, 26, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Grenkowitz, T.; Kassner, U.; Wühle-Demuth, M.; Salewsky, B.; Rosada, A.; Zemojtel, T.; Hopfenmüller, W.; Isermann, B.; Borucki, K.; Heigl, F.; et al. Clinical characterization and mutation spectrum of German patients with familial hypercholesterolemia. Atherosclerosis 2016, 253, 88–93. [Google Scholar] [CrossRef]
- Rabès, J.-P.; Béliard, S.; Carrié, A. Familial hypercholesterolemia. Curr. Opin. Infect. Dis. 2018, 29, 65–71. [Google Scholar] [CrossRef]
- Mickiewicz, A.; Chmara, M.; Futema, M.; Fijalkowski, M.; Chlebus, K.; Galaska, R.; Bandurski, T.; Pajkowski, M.; Zuk, M.; Wasag, B.; et al. Efficacy of clinical diagnostic criteria for familial hypercholesterolemia genetic testing in Poland. Atherosclerosis 2016, 249, 52–58. [Google Scholar] [CrossRef]
- Santos, R.D.; Bourbon, M.; Alonso, R.; Cuevas, A.; Vasques-Cardenas, N.A.; Pereira, A.C.; Merchan, A.; Alves, A.C.; Medeiros, A.M.; Jannes, C.E.; et al. Clinical and molecular aspects of familial hypercholesterolemia in Ibero-American countries. J. Clin. Lipidol. 2017, 11, 160–166. [Google Scholar] [CrossRef]
- Tichý, L.; Fajkusová, L.; Zapletalová, P.; Schwarzová, L.; Vrablík, M.; Freiberger, T. Molecular Genetic Background of an Autosomal Dominant Hypercholesterolemia in the Czech Republic. Physiol. Res. 2017, 66, S47–S54. [Google Scholar] [CrossRef]
- Averna, M.; Cefalù, A.B.; Casula, M.; Noto, D.; Arca, M.; Bertolini, S.; Calandra, S.; Catapano, A.L.; Tarugi, P.; Pellegatta, F.; et al. Familial hypercholesterolemia: The Italian Atherosclerosis Society Network (LIPIGEN). Atheroscler. Suppl. 2017, 29, 11–16. [Google Scholar] [CrossRef]
- Hooper, A.J.; Nguyen, L.T.; Burnett, J.R.; Bates, T.R.; Bell, D.A.; Redgrave, T.G.; Watts, G.F.; van Bockxmeer, F.M. Genetic analysis of familial hypercholesterolaemia in Western Australia. Atherosclerosis 2012, 224, 430–434. [Google Scholar] [CrossRef]
- Jannes, C.E.; Santos, R.D.; de Souza Silva, P.R.; Turolla, L.; Gagliardi, A.C.; Marsiglia, J.D.; Chacra, A.P.; Miname, M.H.; Rocha, V.Z.; Filho, W.S.; et al. Familial hypercholesterolemia in Brazil: Cascade screening program, clinical and genetic aspects. Atherosclerosis 2015, 238, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Latten, M.; Hart, P.; Murray, H.; Bailie, D.A.; Crockard, M.; Lamont, J.; Fitzgerald, P.; Graham, C.A. Genetic diagnosis of familial hypercholesterolaemia using a rapid biochip array assay for 40 common LDLR, APOB and PCSK9 mutations. Atherosclerosis 2016, 254, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Vlad, C.-E.; Foia, L.G.; Popescu, R.; Popa, I.; Aanicai, R.; Reurean-Pintilei, D.; Toma, V.; Florea, L.; Kanbay, M.; Covic, A. Molecular Genetic Approach and Evaluation of Cardiovascular Events in Patients with Clinical Familial Hypercholesterolemia Phenotype from Romania. J. Clin. Med. 2021, 10, 1399. [Google Scholar] [CrossRef] [PubMed]
- De Vries, P.S.; Kavousi, M.; Ligthart, S.; Uitterlinden, A.G.; Hofman, A.; Franco, O.H.; Dehghan, A. Incremental predictive value of 152 single nucleotide polymorphisms in the 10-year risk prediction of incident coronary heart disease: The Rotterdam study. Leuk. Res. 2015, 44, 682–688. [Google Scholar] [CrossRef]
- Cardiero, G.; Ferrandino, M.; Calcaterra, I.L.; Iannuzzo, G.; Di Minno, M.N.D.; Buganza, R.; Guardamagna, O.; Auricchio, R.; Di Taranto, M.D.; Fortunato, G. Impact of 12-SNP and 6-SNP Polygenic Scores on Predisposition to High LDL-Cholesterol Levels in Patients with Familial Hypercholesterolemia. Genes 2024, 15, 462. [Google Scholar] [CrossRef]
- Lima, I.R.; Tada, M.T.; Oliveira, T.G.; Jannes, C.E.; Bensenor, I.; Lotufo, P.A.; Santos, R.D.; Krieger, J.E.; Pereira, A.C. Polygenic risk score for hypercholesterolemia in a Brazilian familial hypercholesterolemia cohort. Atheroscler. Plus 2022, 49, 47–55. [Google Scholar] [CrossRef]
- Leal, L.G.; Hoggart, C.; Jarvelin, M.; Herzig, K.; Sternberg, M.J.E.; David, A. A polygenic biomarker to identify patients with severe hypercholesterolemia of polygenic origin. Mol. Genet. Genom. Med. 2020, 8, e1248. [Google Scholar] [CrossRef]
- Tromp, T.R.; Cupido, A.J.; Reeskamp, L.F.; Stroes, E.S.; Hovingh, G.K.; Defesche, J.C.; Schmidt, A.F.; Zuurbier, L. Assessment of practical applicability and clinical relevance of a commonly used LDL-C polygenic score in patients with severe hypercholesterolemia. Atherosclerosis 2021, 340, 61–67. [Google Scholar] [CrossRef]
- Zadjali, F. LDL cholesterol polygenic score does not reflect polygenic familial hypercholesterolemia in ARABS. Atherosclerosis 2022, 355, 51. [Google Scholar] [CrossRef]
- Wu, H.; Forgetta, V.; Zhou, S.; Bhatnagar, S.R.; Paré, G.; Richards, J.B. Polygenic Risk Score for Low-Density Lipoprotein Cholesterol Is Associated with Risk of Ischemic Heart Disease and Enriches for Individuals with Familial Hypercholesterolemia. Circ. Genom. Precis. Med. 2021, 14, 88–96. [Google Scholar] [CrossRef]
- Martín-Campos, J.M.; Ruiz-Nogales, S.; Ibarretxe, D.; Ortega, E.; Sánchez-Pujol, E.; Royuela-Juncadella, M.; Vila, À.; Guerrero, C.; Zamora, A.; Soler i Ferrer, C.; et al. Polygenic Markers in Patients Diagnosed of Autosomal Dominant Hypercholesterolemia in Catalonia: Distribution of Weighted LDL-c-Raising SNP Scores and Refinement of Variant Selection. Biomedicines 2020, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Francis, G.A.; Brunham, L.R. Association of Monogenic vs. Polygenic Hypercholesterolemia with Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2020, 5, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Weale, M.E.; Riveros-Mckay, F.; Selzam, S.; Seth, P.; Moore, R.; Tarran, W.A.; Gradovich, E.; Giner-Delgado, C.; Palmer, D.; Wells, D.; et al. Validation of an Integrated Risk Tool, Including Polygenic Risk Score, for Atherosclerotic Cardiovascular Disease in Multiple Ethnicities and Ancestries. Am. J. Cardiol. 2021, 148, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Li, X.; DeCastro, M.L.; Cermakova, L.; Sadananda, S.; Jackson, L.M.; Azizi, H.; Mancini, G.J.; Francis, G.A.; Frohlich, J.; et al. Risk of Premature Atherosclerotic Disease in Patients with Monogenic versus Polygenic Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 74, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Sato, T.; Tada, H.; Kannon, T.; Hosomichi, K.; Tsujiguchi, H.; Nakamura, H.; Takamura, M.; Tajima, A.; Kawashiri, M.-A. Polygenic risk scores for low-density lipoprotein cholesterol and familial hypercholesterolemia. J. Hum. Genet. 2021, 66, 1079–1087. [Google Scholar] [CrossRef]
- Cupido, A.J.; Tromp, T.R.; Hovingh, G.K. The clinical applicability of polygenic risk scores for LDL-cholesterol: Considerations, current evidence and future perspectives. Curr. Opin. Infect. Dis. 2021, 32, 112–116. [Google Scholar] [CrossRef]
- Marks, D.; Thorogood, M.; Neil, H.A.W.; Wonderling, D.; Humphries, S.E. Comparing costs and benefits over a 10 year period of strategies for familial hypercholesterolaemia screening. J. Public Health 2003, 25, 47–52. [Google Scholar] [CrossRef]
- Olmastroni, E.; Gazzotti, M.; Arca, M.; Averna, M.; Pirillo, A.; Catapano, A.L.; Casula, M. Twelve Variants Polygenic Score for Low-Density Lipoprotein Cholesterol Distribution in a Large Cohort of Patients with Clinically Diagnosed Familial Hypercholesterolemia with or without Causative Mutations. J. Am. Heart Assoc. 2022, 11, e023668. [Google Scholar] [CrossRef]
- Durst, R.; Ibe, U.K.; Shpitzen, S.; Schurr, D.; Eliav, O.; Futema, M.; Whittall, R.; Szalat, A.; Meiner, V.; Knobler, H.; et al. Molecular genetics of familial hypercholesterolemia in Israel–revisited. Atherosclerosis 2016, 257, 55–63. [Google Scholar] [CrossRef]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic versus Monogenic Causes of Hypercholesterolemia Ascertained Clinically. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef]
- Vanhoye, X.; Bardel, C.; Rimbert, A.; Moulin, P.; Rollat-Farnier, P.-A.; Muntaner, M.; Marmontel, O.; Dumont, S.; Charrière, S.; Cornélis, F.; et al. A new 165-SNP low-density lipoprotein cholesterol polygenic risk score based on next generation sequencing outperforms previously published scores in routine diagnostics of familial hypercholesterolemia. Transl. Res. 2023, 255, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Peloso, G.M.; Zekavat, S.M.; Montasser, M.; Ganna, A.; Chaf, M.; Khera, A.V.; Zhou, W.; Bloom, J.M.; Engreitz, J.M.; et al. Deep-Coverage Whole Genome Sequences and Blood Lipids among 16,324 Individuals. Nat. Commun. 2018, 9, 3391. [Google Scholar] [CrossRef]
- Bolli, A.; Di Domenico, P.; Pastorino, R.; Busby, G.B.; Bottà, G. Risk of Coronary Artery Disease Conferred by Low-Density Lipoprotein Cholesterol Depends on Polygenic Background. Circulation 2021, 143, 1452–1454. [Google Scholar] [CrossRef] [PubMed]
- Damask, A.; Steg, P.G.; Schwartz, G.G.; Szarek, M.; Hagström, E.; Badimon, L.; Chapman, M.J.; Boileau, C.; Tsimikas, S.; Ginsberg, H.N.; et al. Patients with High Genome-Wide Polygenic Risk Scores for Coronary Artery Disease May Receive Greater Clinical Benefit from Alirocumab Treatment in the ODYSSEY OUTCOMES Trial. Circulation 2020, 141, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Young, R.; Stitziel, N.O.; Padmanabhan, S.; Baber, U.; Mehran, R.; Sartori, S.; Fuster, V.; Reilly, D.F.; Butterworth, A.; et al. Polygenic Risk Score Identifies Subgroup with Higher Burden of Atherosclerosis and Greater Relative Benefit from Statin Therapy in the Primary Prevention Setting. Circulation 2017, 135, 2091–2101. [Google Scholar] [CrossRef]
- Marston, N.A.; Kamanu, F.K.; Nordio, F.; Gurmu, Y.; Roselli, C.; Sever, P.S.; Pedersen, T.R.; Keech, A.C.; Wang, H.; Pineda, A.L.; et al. Predicting Benefit from Evolocumab Therapy in Patients with Atherosclerotic Disease Using a Genetic Risk Score. Circulation 2020, 141, 616–623. [Google Scholar] [CrossRef]
- Kullo, I.J.; Jouni, H.; Austin, E.E.; Brown, S.A.; Kruisselbrink, T.M.; Isseh, I.N.; Haddad, R.A.; Marroush, T.S.; Shameer, K.; Olson, J.E.; et al. Incorporating a Genetic Risk Score into Coronary Heart Disease Risk Estimates: Effect on Low-Density Lipoprotein Cholesterol Levels (the MI-GENES Clinical Trial). Circulation 2016, 133, 1181–1188. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC (n = 97) | nFH (DLCN ≤ 8, n = 117) | HC × nFH p-Value | dFH (DLCN > 8, n = 8) | HC × dFH p-Value | nFH × dFH p-Value | |
|---|---|---|---|---|---|---|
| Age (years, median (IQR)) | 47 (43–51) | 50 (46–53) | 0.0012 | 55 (48–57) | 0.0195 | - |
| CHD onset (n, %) | 0 (0.0%) | 117 (100.0%) | 8 (100.0%) | |||
| 21–30 years | 19 (16.2%) | 1 (12.5%) | ||||
| 31–40 years | 68 (58.1%) | 3 (37.5%) | ||||
| 41–50 years | 30 (25.7%) | 4 (50.0%) | ||||
| Male (n, %) | 77 (79.4%) | 96 (82.0%) | - | 5 (62.5%) | - | - |
| Family history of CHD (n, %) | 8 (8.2%) | 41 (35.0%) | <0.0001 | 4 (50.0%) | 0.0004 | - |
| Smoker/ex-smoker (n, %) | 32 (32.9%) | 92 (78.6%) | <0.0001 | 2 (25.0%) | - | 0.0007 |
| Alcohol consumption (n, %) | 36 (37.1%) | 30 (25.6%) | 0.07 | 0 (0.0%) | 0.0344 | 0.10 |
| Arterial hypertension (n, %) | 22 (22.7%) | 55 (47.0%) | 0.0002 | 3 (37.5%) | - | - |
| Body mass index (kg/m2, median (IQR)) | 27.5 (25.4–30.1) | 29.4 (26.2–31.8) | 0.0097 | 30.3 (23.6–31.2) | - | - |
| Lipid-lowering treatment (n, %) | 8 (8.2%) | 80 (68.3%) | <0.0001 | 8 (100.0%) | <0.0001 | 0.059 |
| Total cholesterol (mg/dL, median (IQR)) | 195.5 (173.5–219.1) | 156.7 (134.3–183.8) | <0.0001 | 257.0 (241.1–278.7) | 0.0019 | <0.0001 |
| LDL cholesterol (mg/dL, median (IQR)) | 127.3 (107.8–147.9) | 100.7 (77.7–122.8) | <0.0001 | 191.3 (180.3–222.3) | 0.0001 | <0.0001 |
| eLDL cholesterol 1 (mg/dL, median (IQR)) | 130.7 (108.4–151.9) | 146.9 (113.7–197.4) | 0.0017 | 385.8 (343.0–439.5) | <0.0001 | <0.0001 |
| HDL cholesterol (mg/dL, median (IQR)) | 46.8 (39.0–59.5) | 38.9 (33.8–47.0) | <0.0001 | 37.3 (35.4–52.3) | - | - |
| Non-HDL cholesterol (mg/dL, median (IQR)) | 145.4 (125.4–170.2) | 113.7 (92.1–146.7) | <0.0001 | 222.0 (193.1–232.8) | 0.0018 | <0.0001 |
| Triglycerides (mg/dL, median (IQR)) | 118.5 (79.2–172.2) | 133.2 (104.7–183.1) | 0.0252 | 175.6 (109.1–277.5) | - | - |
| Apo-AI (g/L, median (IQR)) | 1.240 (1.120–1.390) | 1.140 (0.974–1.310) | 0.0009 | 1.215 (0.976–1.390) | - | - |
| Apo-AII (g/L, median (IQR)) | 0.353 (0.318–0.411) | 0.316 (0.282–0.353) | <0.0001 | 0.325 (0.297–0.384) | - | - |
| Apo-B (g/L, median (IQR)) | 0.868 (0.749–0.995) | 0.811 (0.656–0.945) | 0.0238 | 1.260 (1.155–1.390) | 0.0005 | 0.0002 |
| Apo-E (g/L, median (IQR)) | 0.039 (0.034–0.047) | 0.036 (0.032–0.043) | 0.0274 | 0.049 (0.040–0.058) | 0.0461 | 0.0082 |
| Lipoprotein(a) (mg/dL, median (IQR)) | 9.7 (9.7–16.0) | 9.7 (9.7–29.8) | - | 9.8 (9.7–24.9) | - | - |
| DLCN score 2 (n, (IQR)) | 0 (0–1) | 3 (2–5) | <0.0001 | 11 (10–11) | <0.0001 | <0.0001 |
| Unlikely FH (n, %) | 92 (94.8%) | 40 (34.2%) | 0 (0.0%) | |||
| Possible FH (n, %) | 5 (5.2%) | 53 (45.3%) | 0 (0.0%) | |||
| Probable FH (n, %) | 0 (0.0%) | 24 (20.5%) | 0 (0.0%) | |||
| Definite FH (n, %) | 0 (0.0%) | 0 (0.0%) | 8 (100.0%) |
| Reference SNP Cluster ID | Gene | Minor Allele (m) | Common Allele (M) | Contrib. to aPRS (m/M) | Contrib. to wPRS (m/M) | HC (n = 97) | nFH (n = 117) | dFH (n = 8) |
|---|---|---|---|---|---|---|---|---|
| Minor Allele Frequency | ||||||||
| rs6511720 | LDLR | T 1 | G * | 0/+1 | 0/+0.180 | 0.118 | 0.068 | 0.000 |
| rs629301 | CELSR2 | G 1 | T * | 0/+1 | 0/+0.146 | 0.221 | 0.196 | 0.000 |
| rs1367117 | APOB | A * | G | +1/0 | +0.105/0 | 0.345 | 0.303 | 0.312 |
| rs4299376 | ABCG8 | G * | T | +1/0 | +0.071/0 | 0.350 | 0.359 | 0.250 |
| rs1800562 | HFE | A 1 | G * | 0/+1 | 0/+0.057 | 0.031 | 0.004 | 0.000 |
| rs2479409 | PCSK9 | G * | A | +1/0 | +0.052/0 | 0.350 | 0.363 | 0.437 |
| rs429358 | APOE | C | T | - | - | 0.118 | 0.128 | 0.062 |
| rs7412 | APOE | T | C | - | - | 0.072 | 0.051 | 0.000 |
| APOE genotype | APOE genotype frequency | |||||||
| ε2ε2 | - | - | −2 | −0.800 | 0.000 | 0.000 | 0.000 | |
| ε2ε3 | - | - | −1 | −0.400 | 0.103 | 0.102 | 0.000 | |
| ε2ε4 | - | - | 0 | −0.270 | 0.041 | 0.000 | 0.000 | |
| ε3ε3 | - | - | 0 | 0 | 0.660 | 0.719 | 0.875 | |
| ε3ε4 * | - | - | +1 | +0.130 | 0.196 | 0.162 | 0.125 | |
| ε4ε4 * | - | - | +2 | +0.260 | 0.000 | 0.017 | 0.000 | |
| 8-SNP LDLC polygenic risk score (PRS) | Median PRS (IQR) | |||||||
| Additive (a)PRS | 7.0 (7.0–9.0) | 8.0 (6.7–8.2) | 8.0 (7.5–9.0) | |||||
| Weighted (w)PRS | 0.818 (0.661–0.949) | 0.837 (0.691–0.953) | 0.906 (0.853–1.028) | |||||
| Variable | Measurement Level | Standardized β Coefficient or Odds Ratio with (95% CI) and p-Value | |||
|---|---|---|---|---|---|
| Combined Group (n = 222) | HC (n = 97) | PCHD (n = 125) | PCHD nFH (n = 117) | ||
| Age 1 | ≤45 years | Ref | Ref | Ref | Ref |
| >45 years | 1.01 (0.76–1.34), p = 0.933 | 0.82 (0.54–1.25), p = 0.360 | 1.14 (0.75–1.73), p = 0.526 | 1.09 (0.71–1.68), p = 0.674 | |
| Sex 1 | Female | Ref | Ref | Ref | Ref |
| Male | 1.05 (0.76–1.46), p = 0.748 | 1.15 (0.71–1.85), p = 0.566 | 0.96 (0.61–1.51), p = 0.874 | 0.96 (0.59–1.55), p = 0.871 | |
| Family history of CHD 1 | No | Ref | Ref | Ref | Ref |
| Yes | 1.27 (0.91–1.77), p = 0.138 | 1.21 (0.55–2.68), p = 0.618 | 1.19 (0.82–1.74), p = 0.340 | 1.15 (0.78–1.70), p = 0.467 | |
| Arterial hypertension 1 | No | Ref | Ref | Ref | Ref |
| Yes | 1.17 (0.88–1.56), p = 0.253 | 1.30 (0.77–2.19), p = 0.309 | 1.04 (0.73–1.48), p = 0.813 | 1.09 (0.75–1.57), p = 0.646 | |
| BMI 2 | SD variation | 0.15 (0.02–0.28), p = 0.018 | 0.15 (−0.05; 0.35), p = 0.142 | 0.13 (−0.03; 0.31), p = 0.123 | 0.16 (−0.01; 0.34), p = 0.075 |
| DLCN score 2 | SD variation | 0.16 (0.03–0.29), p = 0.015 | 0.19 (0.00–0.39), p = 0.051 | 0.13 (−0.04; 0.30), p = 0.146 | 0.05 (−0.13; 0.23), p = 0.590 |
| eLDLC 2 | SD variation | 0.20 (0.07–0.33), p = 0.002 | 0.25 (0.06–0.45), p = 0.011 | 0.18 (0.01–0.36), p = 0.036 | 0.14 (−0.04; 0.32), p = 0.133 |
| eLDLC 1 | ≤190 mg/dL | Ref | Ref | Ref | Ref |
| >190 mg/dL | 1.36 (0.96–1.92), p = 0.070 | 1.98 (0.63–6.17), p = 0.187 | 1.20 (0.82–1.75), p = 0.339 | 1.09 (0.73–1.62), p = 0.667 | |
| HDLC 2 | SD variation | −0.14 (−0.27; −0.01), p = 0.029 | −0.06 (−0.26; 0.14), p = 0.540 | −0.18 (−0.35; 0), p = 0.041 | −0.21 (−0.39; −0.03), p = 0.023 |
| Lipoprotein(a) 2 | SD variation | 0.05 (−0.08; 0.18), p = 0.463 | −0.02 (−0.23; 0.17), p = 0.791 | 0.07 (−0.10; 0.25), p = 0.421 | 0.07 (−0.10; 0.26), p = 0.416 |
| Variable | Measurement Level | Unadjusted OR (95% CI) | p-Value |
|---|---|---|---|
| Age | ≤45 years | Ref | |
| >45 years | 2.75 (1.54–4.93) | 0.0005 | |
| Sex | Female | Ref | |
| Male | 1.09 (0.56–2.12) | 0.792 | |
| Family history of CHD | No | Ref | |
| Yes | 6.25 (2.78–14.07) | <0.0001 | |
| Arterial hypertension | No | Ref | |
| Yes | 2.95 (1.63–5.33) | 0.0002 | |
| Body mass index | kg/m2 | 1.08 (1.01–1.15) | 0.018 |
| Body mass index 1 | 1st quartile | Ref | |
| 2nd quartile | 0.84 (0.40–1.78) | 0.655 | |
| 3rd quartile | 1.68 (0.78–3.61) | 0.181 | |
| 4th quartile | 2.38 (1.08–5.21) | 0.028 | |
| Smoking | No | Ref | |
| Current/past | 6.16 (3.42–11.07) | <0.0001 | |
| Alcohol consumption | No | Ref | |
| Yes | 0.53 (0.30–0.95) | 0.034 | |
| DLCN score | Score unit | 12.61 (6.05–26.28) | <0.0001 |
| eLDLC | mg/dL | 1.011 (1.006–1.017) | <0.0001 |
| eLDLC | ≤190 mg/dL | Ref | |
| >190 mg/dL | 8.23 (3.33–20.34) | <0.0001 | |
| HDLC | mg/dL | 0.948 (0.927–0.970) | <0.0001 |
| Lipoprotein(a) | mg/dL | 1.016 (1.002–1.030) | 0.016 |
| Triglyceride | mg/dL | 1.00 (0.99–1.01) | 0.971 |
| aPRS | SD | 1.14 (0.87–1.49) | 0.317 |
| wPRS | SD | 1.22 (0.93–1.59) | 0.142 |
| Variable | Measurement Level | Reduced Model | Complete Model | ||
|---|---|---|---|---|---|
| Odds Ratio (95% CI) | p-Value | Odds Ratio (95% CI) | p-Value | ||
| Alcohol consumption | No | Ref | Ref | ||
| Yes | 0.25 (0.11–0.56) | 0.0006 | 0.29 (0.13–0.67) | 0.0036 | |
| Arterial hypertension | No | Ref | Ref | ||
| Yes | 2.98 (1.44–6.17) | 0.0032 | 2.42 (1.08–5.41) | 0.0304 | |
| BMI | kg/m2 | 1.07 (0.98–1.16) | 0.0938 | - | - |
| Family history of CHD | No | Ref | Ref | ||
| Yes | 10.35 (3.93–27.24) | <0.0001 | 10.01 (3.33–30.09) | <0.0001 | |
| Smoking (past/current) | No | Ref | Ref | ||
| Yes | 10.09 (4.80–21.22) | <0.0001 | 10.86 (4.83–24.41) | <0.0001 | |
| eLDLC | mg/dL | - | - | 1.012 (1.005–1.020) | 0.0012 |
| HDLC | mg/dL | - | - | 0.949 (0.920–0.979) | 0.0009 |
| Lipoprotein(a) | mg/dL | - | - | 1.020 (1.001–1.039) | 0.0352 |
| Model performance | |||||
| Overall model p value | <0.0001 | <0.0001 | |||
| Cox and Snell R2 | 0.34 | 0.43 | |||
| AUC value (95% CI) | 0.855 (0.802–0.899) | 0.895 (0.847–0.932) | |||
| Specificity | 75.2% | 78.3% | |||
| Sensitivity | 81.6% | 84.8% | |||
| Accuracy | 78.8% | 81.9% | |||
| Training Group | Testing Group | AUC | ||||||
|---|---|---|---|---|---|---|---|---|
| CC% | Comparison | CC% | Comparison | Median | IQR | Range | Comparison | |
| Model 1 | 83.3 | p = 0.02 | 82.6 | p = 0.07 | 0.898 | 0.893–0.903 | 0.854–0.924 | p = 0.34 |
| Model 2 | 84.1 | 82.6 | 0.898 | 0.892–0.906 | 0.864–0.922 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mănescu, I.B.; Gabor, M.R.; Moldovan, G.V.; Hadadi, L.; Huțanu, A.; Bănescu, C.; Dobreanu, M. An 8-SNP LDL Cholesterol Polygenic Score: Associations with Cardiovascular Risk Traits, Familial Hypercholesterolemia Phenotype, and Premature Coronary Heart Disease in Central Romania. Int. J. Mol. Sci. 2024, 25, 10038. https://doi.org/10.3390/ijms251810038
Mănescu IB, Gabor MR, Moldovan GV, Hadadi L, Huțanu A, Bănescu C, Dobreanu M. An 8-SNP LDL Cholesterol Polygenic Score: Associations with Cardiovascular Risk Traits, Familial Hypercholesterolemia Phenotype, and Premature Coronary Heart Disease in Central Romania. International Journal of Molecular Sciences. 2024; 25(18):10038. https://doi.org/10.3390/ijms251810038
Chicago/Turabian StyleMănescu, Ion Bogdan, Manuela Rozalia Gabor, George Valeriu Moldovan, László Hadadi, Adina Huțanu, Claudia Bănescu, and Minodora Dobreanu. 2024. "An 8-SNP LDL Cholesterol Polygenic Score: Associations with Cardiovascular Risk Traits, Familial Hypercholesterolemia Phenotype, and Premature Coronary Heart Disease in Central Romania" International Journal of Molecular Sciences 25, no. 18: 10038. https://doi.org/10.3390/ijms251810038