
Licochalcone D from Glycyrrhiza uralensis Improves High-Glucose-Induced Insulin Resistance in Hepatocytes


Abstract
:1. Introduction
2. Results
2.1. Determination of LicoD Contents in G. uralensis
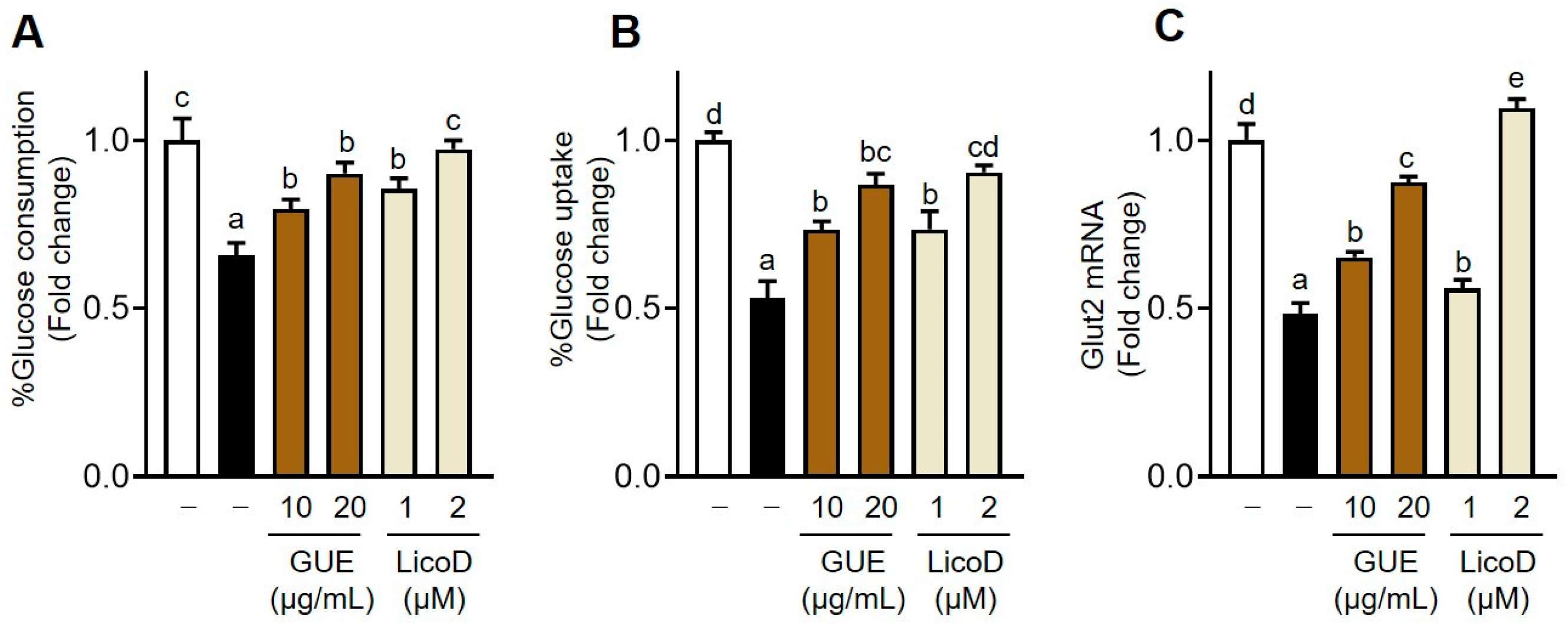
2.2. Regulation of Glucose Metabolism by GUE or LicoD in IR Hepatocytes
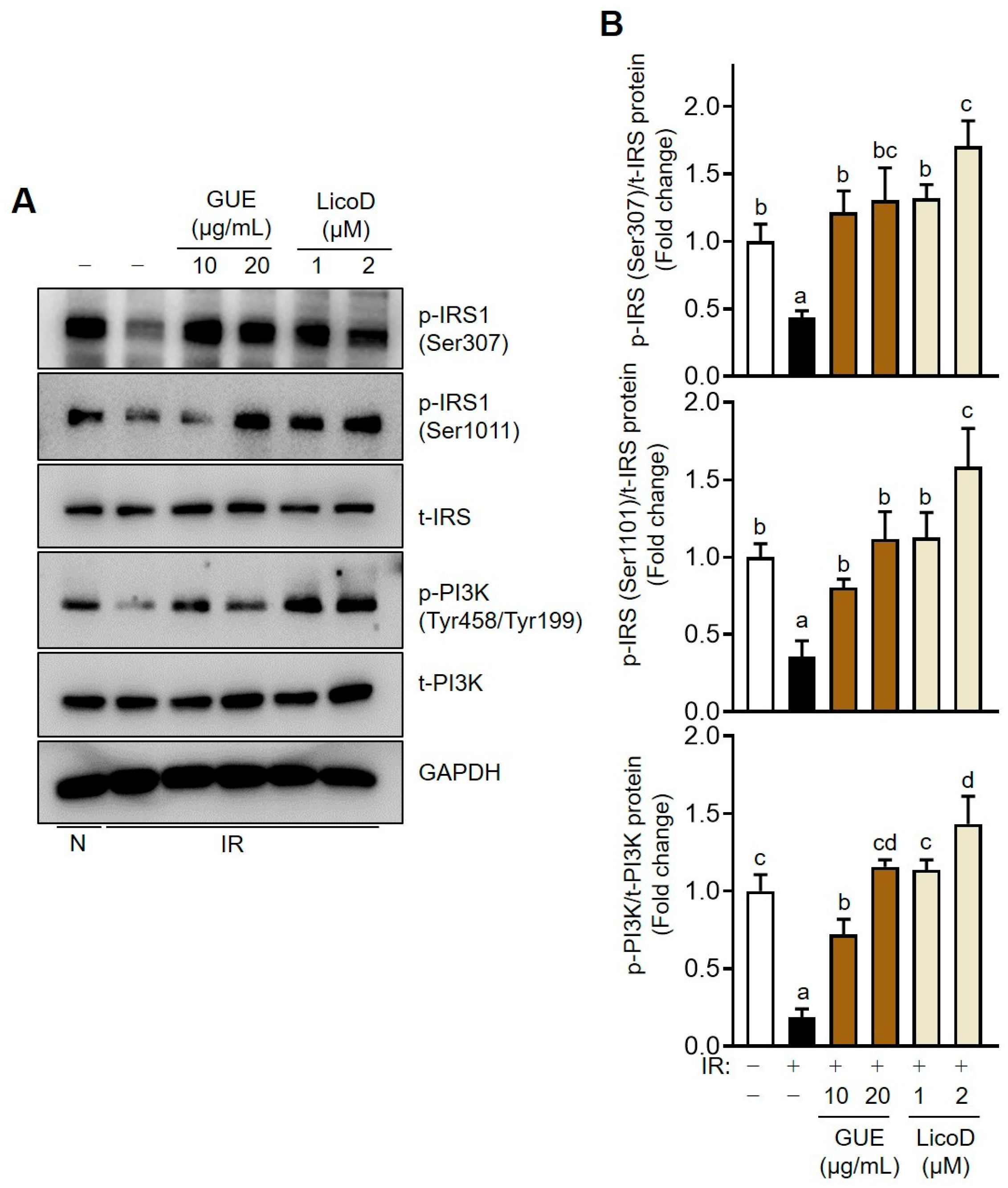
2.3. Enhancement of the IRS/PI3K Pathway by GUE or LicoD in IR Hepatocytes
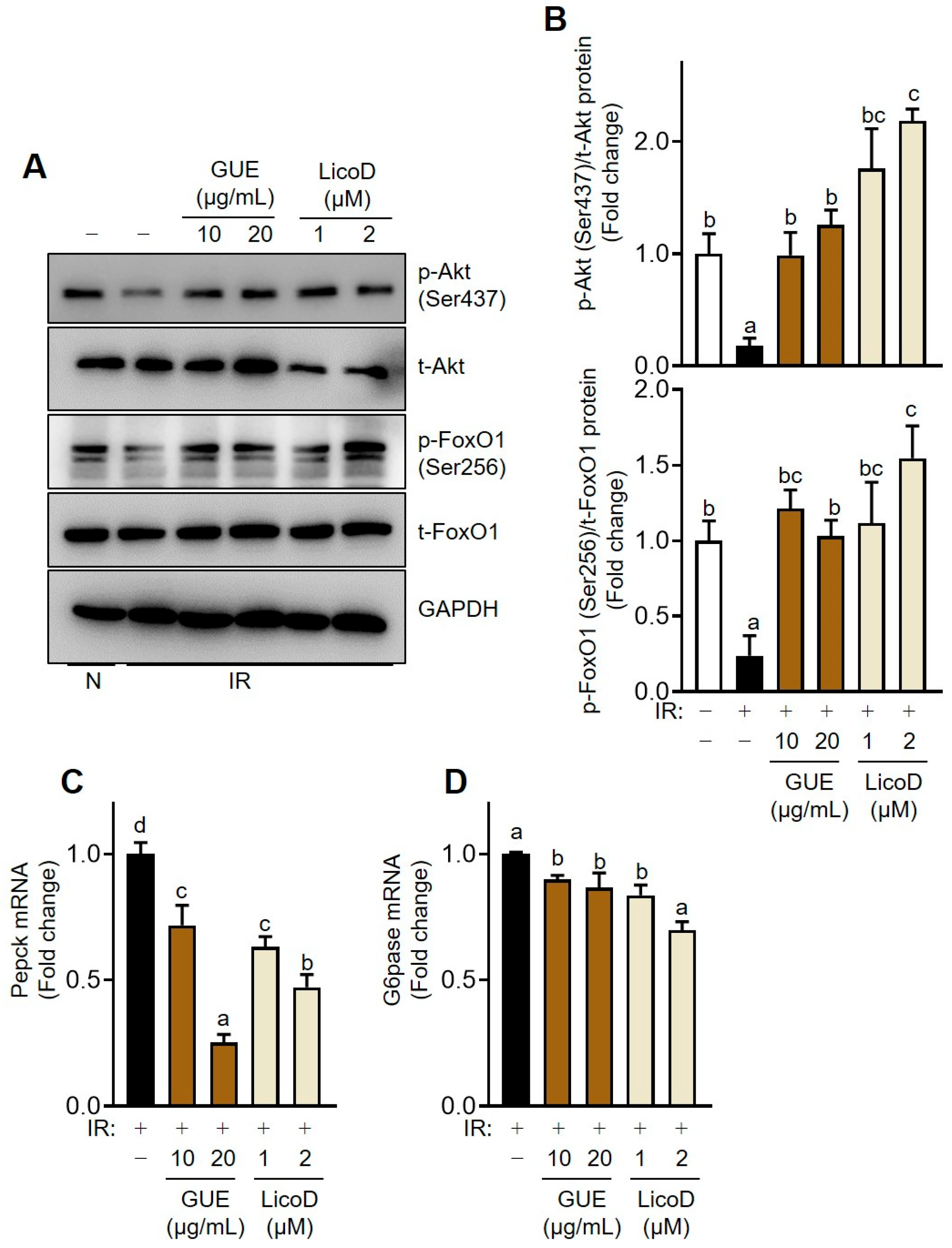
2.4. Inhibition of Akt-Mediated Gluconeogenesis by GUE or LicoD in IR Hepatocytes
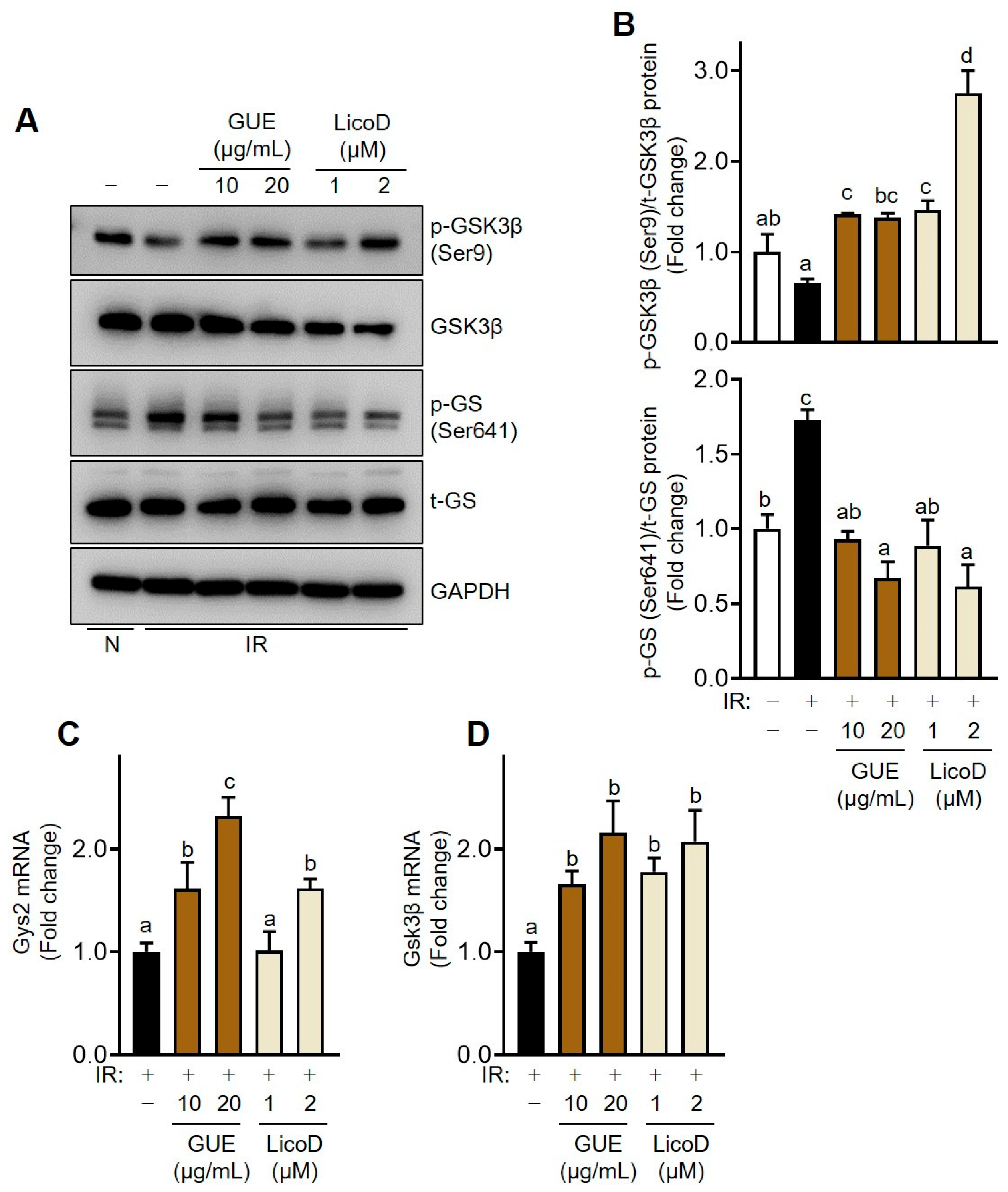
2.5. Enhancement of Glycogen Synthesis by GUE or LicoD in IR Hepatocytes
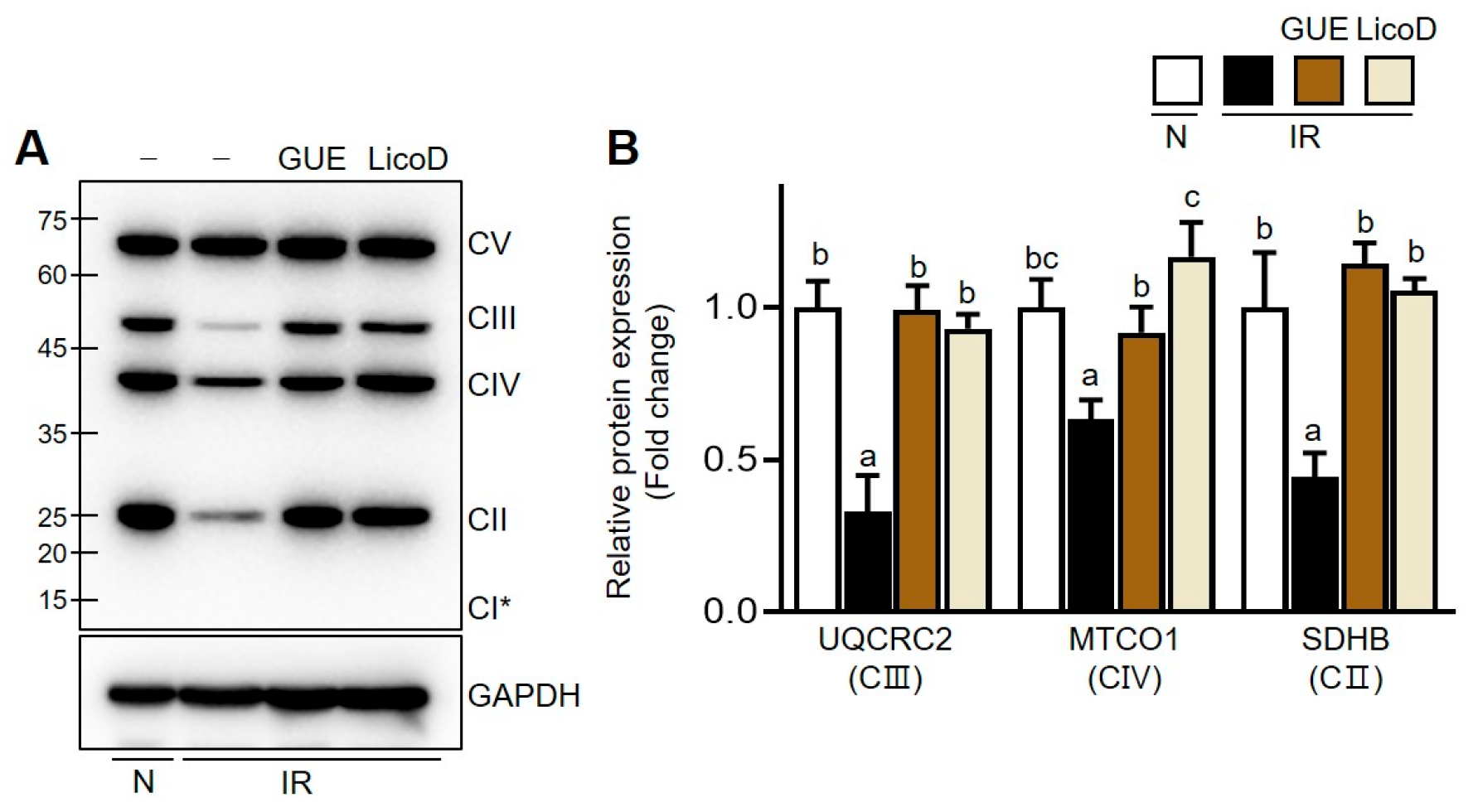
2.6. Improvement of the PPARα/PGC1α Pathway by GUE or LicoD in IR Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Extraction and Identification of LicoD from G. uralensis
4.3. Cell Culture and Treatments
4.4. Cell Viability Assay
4.5. Glucose Consumption Assay
4.6. Glucose Uptake Assay
4.7. Quantitative Real-Time Polymerase Chain Reaction
4.8. Western Blot Assay
4.9. Mitochondrial DNA Quantification
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Frankowski, R.; Kobierecki, M.; Wittczak, A.; Różycka-Kosmalska, M.; Pietras, T.; Sipowicz, K.; Kosmalski, M. Type 2 Diabetes Mellitus, Non-Alcoholic Fatty Liver Disease, and Metabolic Repercussions: The Vicious Cycle and Its Interplay with Inflammation. Int. J. Mol. Sci. 2023, 24, 9677. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei-Malazy, O.; Larijani, B.; Abdollahi, M. Targeting metabolic disorders by natural products. J. Diabetes Metab. Disord. 2015, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G., Jr.; Seeley, R.J.; Schwartz, M.W. Insulin Activation of Phosphatidylinositol 3-Kinase in the Hypothalamic Arcuate Nucleus: A Key Mediator of Insulin-Induced Anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef]
- Feng, Y.; Ren, Y.; Zhang, X.; Yang, S.; Jiao, Q.; Li, Q.; Jiang, W. Metabolites of traditional Chinese medicine targeting PI3K/AKT signaling pathway for hypoglycemic effect in type 2 diabetes. Front. Pharmacol. 2024, 15, 1373711. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483. [Google Scholar] [CrossRef]
- Teli, D.M.; Gajjar, A.K. Glycogen synthase kinase-3: A potential target for diabetes. Bioorganic Med. Chem. 2023, 92, 117406. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Di, L.J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Nunnari, J. Mitochondria at the crossroads of health and disease. Cell 2024, 187, 2601–2627. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Apostolova, N.; Diaz-Rua, R.; Muntane, J.; Victor, V.M. Mitochondria and T2D: Role of autophagy, ER stress, and inflammasome. Trends Endocrinol. Metab. 2020, 31, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E.; Ragavan, M.; Palavicini, J.P.; Fourcaudot, M.; Bakewell, T.M.; Valdez, I.A.; Ayala, I.; Jin, E.S.; Madesh, M.; Han, X.; et al. Insulin resistance is mechanistically linked to hepatic mitochondrial remodeling in non-alcoholic fatty liver disease. Mol. Metab. 2021, 45, 101154. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-a.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Collins, Y.; Logan, A.; Murphy, M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. 2012, 23, 429–434. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, M. Chemical analysis of the Chinese herbal medicine Gan-Cao (licorice). J. Chromatogr. A 2009, 1216, 1954–1969. [Google Scholar] [CrossRef]
- So-Young, L.; So-Lim, P.; Sung-Hun, Y.; Young-Do, N.; Seong-Il, L. Quality Characteristics of Low-Salt Gochujang Added with Glycyrrhiza uralensis and Brassica juncea. Prev. Nutr. Food Sci. 2011, 16, 348–356. [Google Scholar]
- Liao, W.C.; Lin, Y.-H.; Chang, T.-M.; Huang, W.-Y. Identification of two licorice species, Glycyrrhiza uralensis and Glycyrrhiza glabra, based on separation and identification of their bioactive components. Food Chem. 2012, 132, 2188–2193. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Li, Y.-J.; Zheng, Y.-F.; Li, P. Antioxidant and anti-inflammatory activities of six flavonoids separated from licorice. Food Chem. 2013, 141, 1063–1071. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Dong, C.; Zhuang, Y.; Hao, G.; Wang, F. Licochalcone D exhibits cytotoxicity in breast cancer cells and enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis through upregulation of death receptor 5. J. Biochem. Mol. Toxicol. 2024, 38, e23757. [Google Scholar] [CrossRef] [PubMed]
- Yudhani, R.D.; Sari, Y.; Nugrahaningsih, D.A.A.; Sholikhah, E.N.; Rochmanti, M.; Purba, A.K.R.; Khotimah, H.; Nugrahenny, D.; Mustofa, M. In Vitro Insulin Resistance Model: A Recent Update. J. Obes. 2023, 2023, 1964732. [Google Scholar] [CrossRef] [PubMed]
- Guerre-Millo, M.; Rouault, C.; Poulain, P.; André, J.; Poitout, V.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.-C.; Reach, G.; Staels, B. PPAR-α–null mice are protected from high-fat diet–induced insulin resistance. Diabetes 2001, 50, 2809–2814. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [PubMed]
- Lee, H.E.; Yang, G.; Han, S.-H.; Lee, J.-H.; An, T.-J.; Jang, J.-K.; Lee, J.Y. Anti-obesity potential of Glycyrrhiza uralensis and licochalcone A through induction of adipocyte browning. Biochem. Biophys. Res. Commun. 2018, 503, 2117–2123. [Google Scholar] [CrossRef]
- Teng, H.; Chen, M.; Zou, A.; Jiang, H.; Han, J.; Sun, L.; Feng, C.; Liu, J. Hepatoprotective effects of licochalcone B on carbon tetrachloride-induced liver toxicity in mice. Iran. J. Basic. Med. Sci. 2016, 19, 910. [Google Scholar]
- Ahmad, K.; Lee, E.J.; Ali, S.; Han, K.S.; Hur, S.J.; Lim, J.H.; Choi, I. Licochalcone A and B enhance muscle proliferation and differentiation by regulating Myostatin. Phytomedicine 2024, 125, 155350. [Google Scholar] [CrossRef]
- Mae, T.; Kishida, H.; Nishiyama, T.; Tsukagawa, M.; Konishi, E.; Kuroda, M.; Mimaki, Y.; Sashida, Y.; Takahashi, K.; Kawada, T.; et al. A licorice ethanolic extract with peroxisome proliferator-activated receptor-gamma ligand-binding activity affects diabetes in KK-Ay mice, abdominal obesity in diet-induced obese C57BL mice and hypertension in spontaneously hypertensive rats. J. Nutr. 2003, 133, 3369–3377. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Zhang, L.; Chen, Y.; Wu, T.; Liu, R.; Sui, W.; Zhu, Q.; Zhang, M. Licorice extract ameliorates hyperglycemia through reshaping gut microbiota structure and inhibiting TLR4/NF-κB signaling pathway in type 2 diabetic mice. Food Res. Int. 2022, 153, 110945. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Małkowska, P. Positive Effects of Physical Activity on Insulin Signaling. Curr. Issues Mol. Biol. 2024, 46, 5467–5487. [Google Scholar] [CrossRef] [PubMed]
- Onyango, A.N. Excessive gluconeogenesis causes the hepatic insulin resistance paradox and its sequelae. Heliyon 2022, 8, e12294. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, H.; Deng, J.; Fan, D. Ginsenoside Rg5 Improves Insulin Resistance and Mitochondrial Biogenesis of Liver via Regulation of the Sirt1/PGC-1α Signaling Pathway in db/db Mice. J. Agric. Food Chem. 2021, 69, 8428–8439. [Google Scholar] [CrossRef]
- Ye, X.; Chen, W.; Huang, X.F.; Yan, F.J.; Deng, S.G.; Zheng, X.D.; Shan, P.F. Anti-diabetic effect of anthocyanin cyanidin-3-O-glucoside: Data from insulin resistant hepatocyte and diabetic mouse. Nutr. Diabetes 2024, 14, 7. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, Y.; Li, P.F. PPARα: An emerging target of metabolic syndrome, neurodegenerative and cardiovascular diseases. Front. Endocrinol. 2022, 13, 1074911. [Google Scholar] [CrossRef]
- Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef]
- Lalloyer, F.; Vandewalle, B.; Percevault, F.; Torpier, G.; Kerr-Conte, J.; Oosterveer, M.; Paumelle, R.J.; Fruchart, J.-C.; Kuipers, F.; Pattou, F. Peroxisome proliferator–activated receptor α improves pancreatic adaptation to insulin resistance in obese mice and reduces lipotoxicity in human islets. Diabetes 2006, 55, 1605–1613. [Google Scholar] [CrossRef]
- Rodríguez-Vilarrupla, A.; Laviña, B.; García-Calderó, H.; Russo, L.; Rosado, E.; Roglans, N.; Bosch, J.; García-Pagán, J.C. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–1039. [Google Scholar] [CrossRef]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.G. Peroxisome proliferator activated receptor-alpha (PPARα) and PPAR gamma coactivator-1alpha (PGC-1α) regulation of cardiac metabolism in diabetes. Pediatr. Cardiol. 2011, 32, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Jong-Hyun, H.; Kyong-Haeng, L.; Kwang-Won, Y. Characterization of the Immunologically Active Components of Glycyrrhiza uralensis Prepared as Herbal Kimchi. Prev. Nutr. Food Sci. 2003, 8, 29–35. [Google Scholar]
- Jung, J.C.; Lee, Y.H.; Kim, S.H.; Kim, K.J.; Kim, K.M.; Oh, S.; Jung, Y.S. Hepatoprotective effect of licorice, the root of Glycyrrhiza uralensis Fischer, in alcohol-induced fatty liver disease. BMC Complement. Altern. Med. 2016, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.K.; Han, Y.; Park, H.J.; Shin, M.R.; Roh, S.S.; Kwon, E.Y. The Synergistic Action of Metformin and Glycyrrhiza uralensis Fischer Extract Alleviates Metabolic Disorders in Mice with Diet-Induced Obesity. Int. J. Mol. Sci. 2023, 24, 936. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.H.; Paul, C.R.; Kuo, C.H.; Chang, Y.H.; Chen, W.S.T.; Ho, T.J.; Day, C.H.; Velmurugan, B.K.; Tsai, Y.; Huang, C.Y. Glycyrrhiza uralensis root extract ameliorates high glucose-induced renal proximal tubular fibrosis by attenuating tubular epithelial-myofibroblast transdifferentiation by targeting TGF-β1/Smad/Stat3 pathway. J. Food Biochem. 2022, 46, e14041. [Google Scholar] [CrossRef]
- Park, S.K.; Lee, J.; Park, S.H.; Lee, Y.G. Anti-obesity effects of Glycyrrhiza uralensis ethanol extract on the inhibition of 3T3-L1 adipocyte differentiation in high-fat diet-induced C57BL/6J mice. Food Sci. Preserv. 2023, 30, 716–728. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample * | Contents ** (µg/100 mg dry wt.) |
|---|---|
| GUE | 8.61 ± 0.51 |
| GUH | 0.85 ± 0.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.G.; Lee, H.M.; Hwang, J.-T.; Choi, H.-K. Licochalcone D from Glycyrrhiza uralensis Improves High-Glucose-Induced Insulin Resistance in Hepatocytes. Int. J. Mol. Sci. 2024, 25, 10066. https://doi.org/10.3390/ijms251810066
Lee YG, Lee HM, Hwang J-T, Choi H-K. Licochalcone D from Glycyrrhiza uralensis Improves High-Glucose-Induced Insulin Resistance in Hepatocytes. International Journal of Molecular Sciences. 2024; 25(18):10066. https://doi.org/10.3390/ijms251810066
Chicago/Turabian StyleLee, Yu Geon, Hee Min Lee, Jin-Taek Hwang, and Hyo-Kyoung Choi. 2024. "Licochalcone D from Glycyrrhiza uralensis Improves High-Glucose-Induced Insulin Resistance in Hepatocytes" International Journal of Molecular Sciences 25, no. 18: 10066. https://doi.org/10.3390/ijms251810066