
Augmented Global Protein Acetylation Diminishes Cell Growth and Migration of Cholangiocarcinoma Cells

 , , , , , ,
, , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
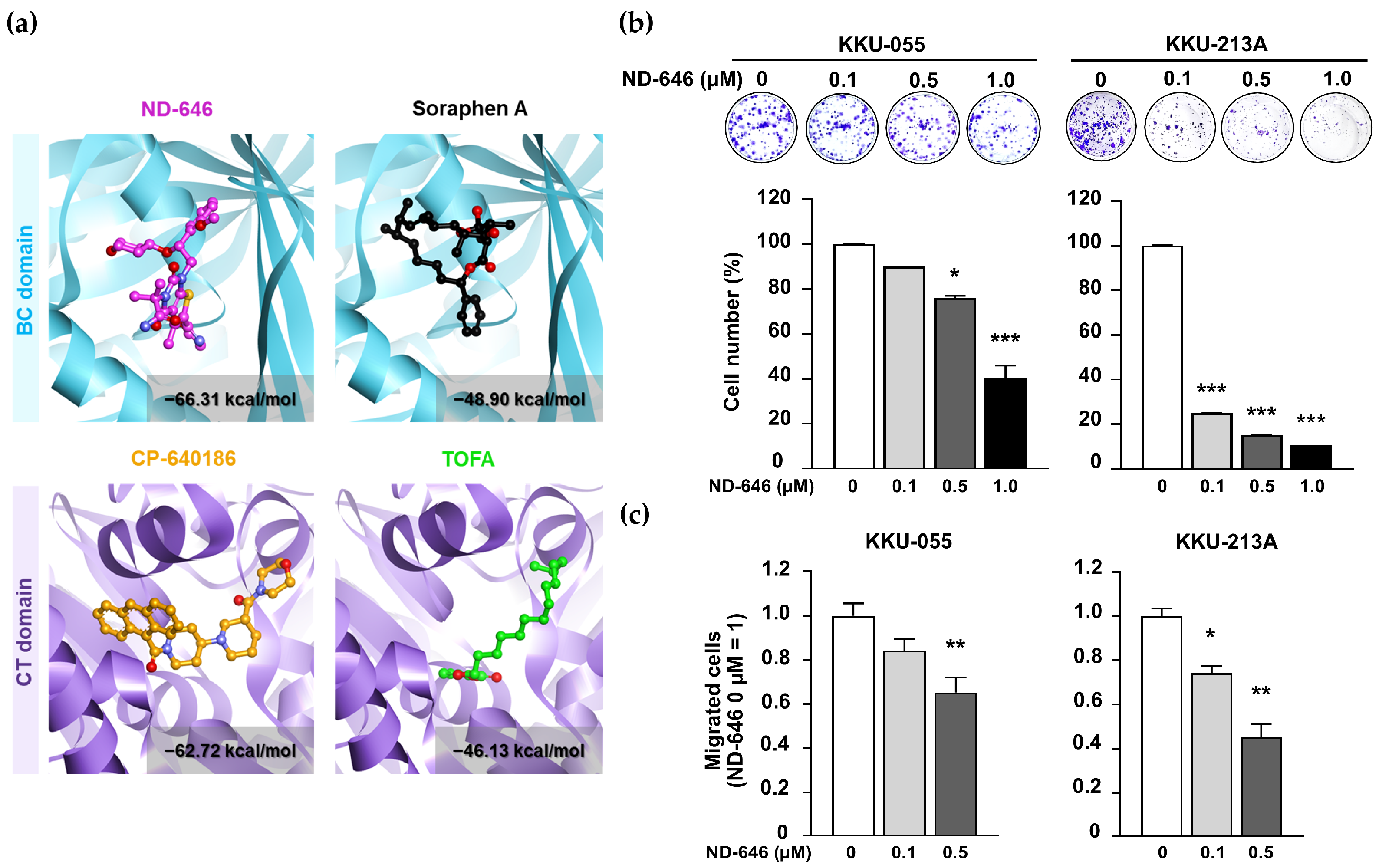
2.1. ND-646 Suppressed CCA Cell Proliferation and Migration
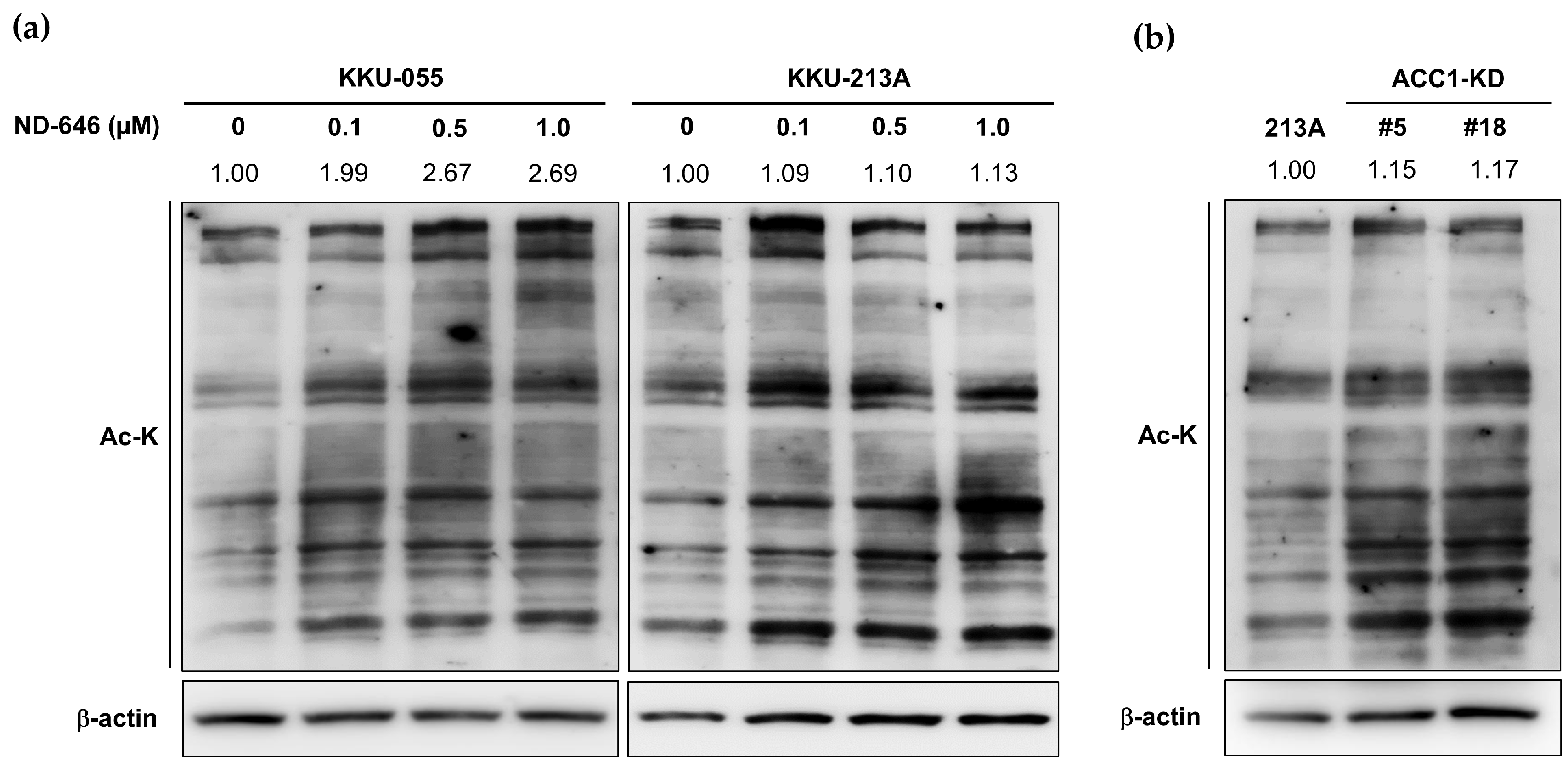
2.2. Increased Total Protein Acetylation in ACC1 Inhibitory Cells
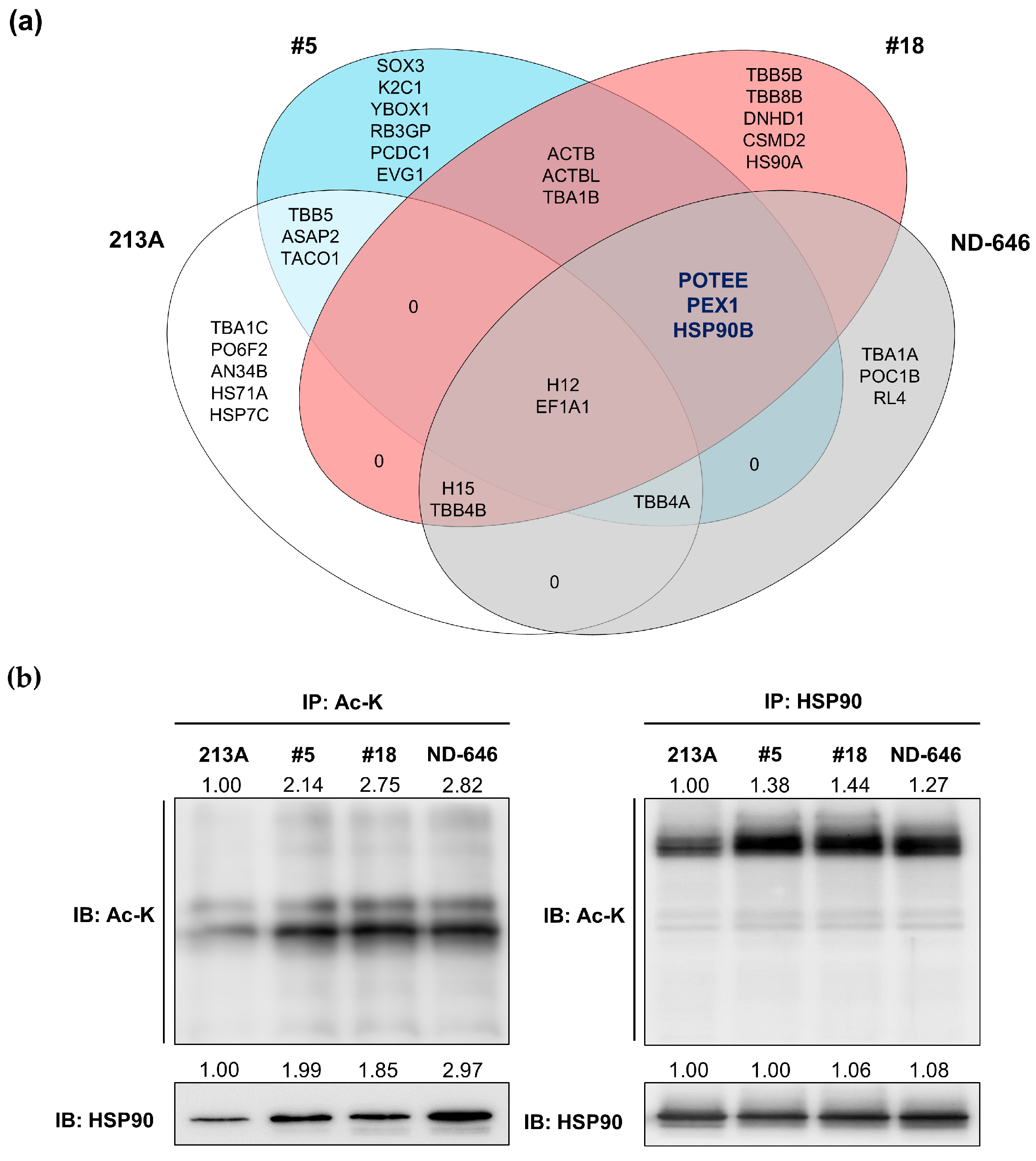
2.3. HSP90B, PEX1, and POTEE Were Acetylated in ACC1-Deficient Cells
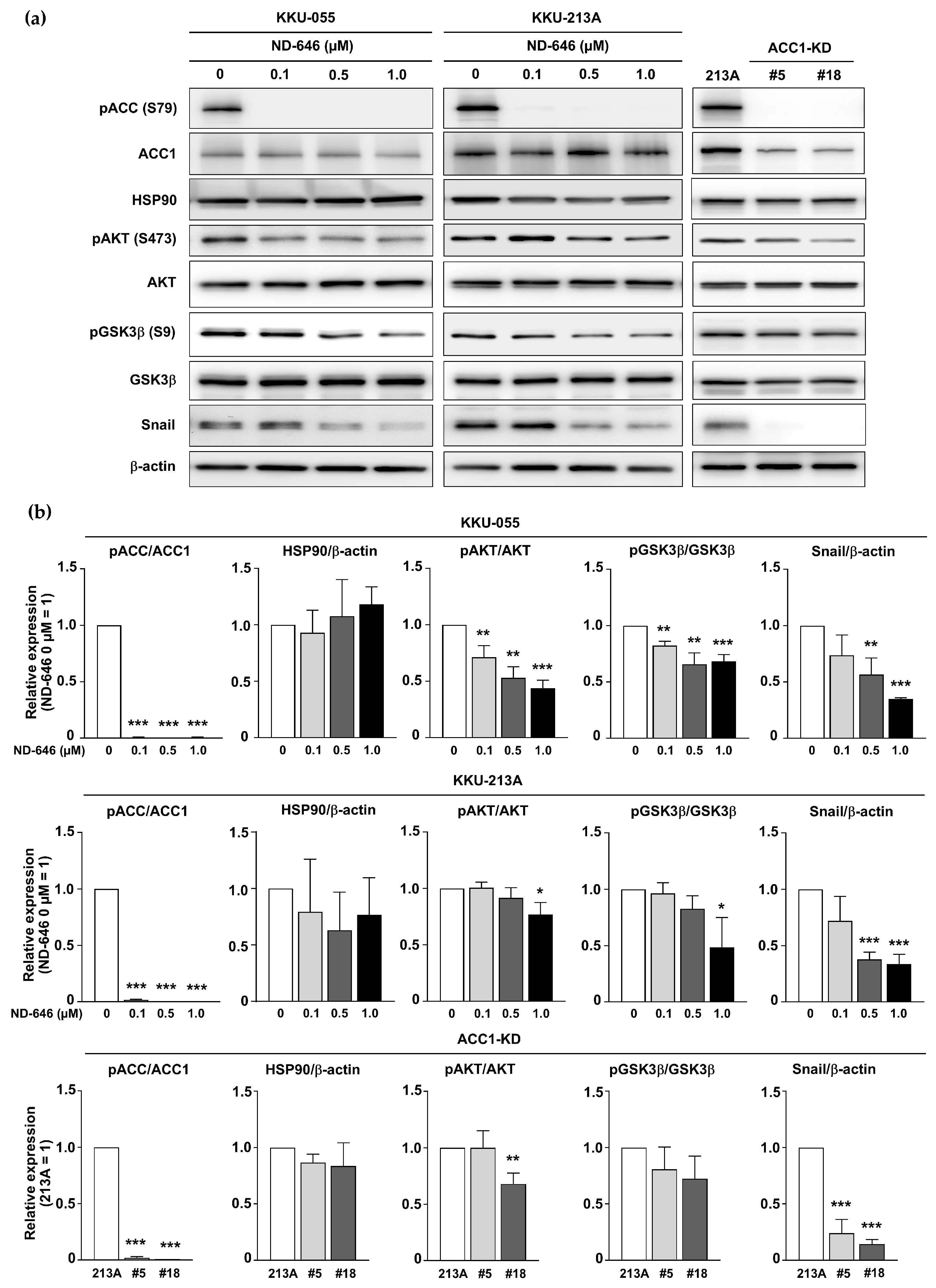
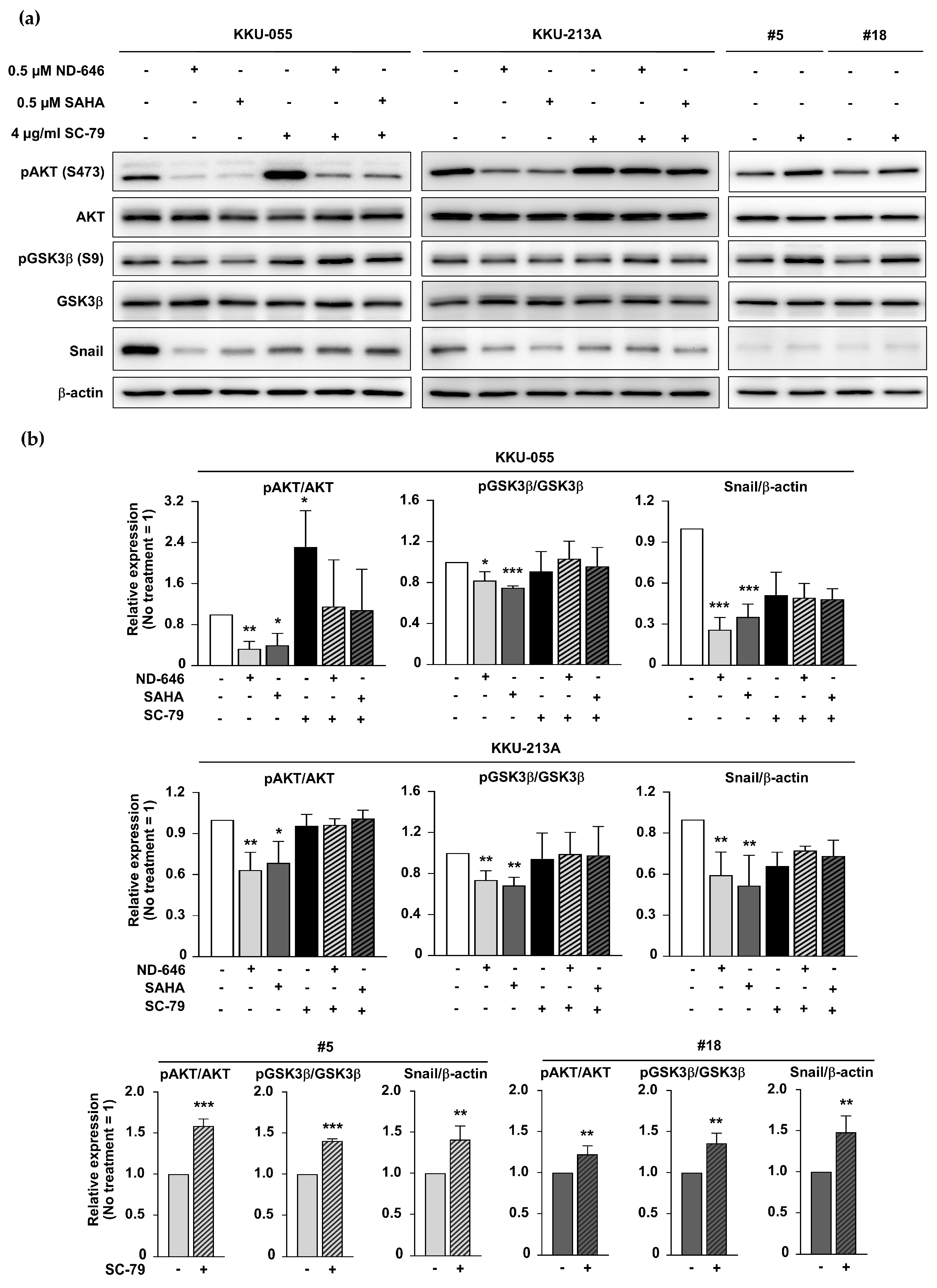
2.4. Inhibition of ACC1 Affected the AKT/GSK3β/Snail Axis
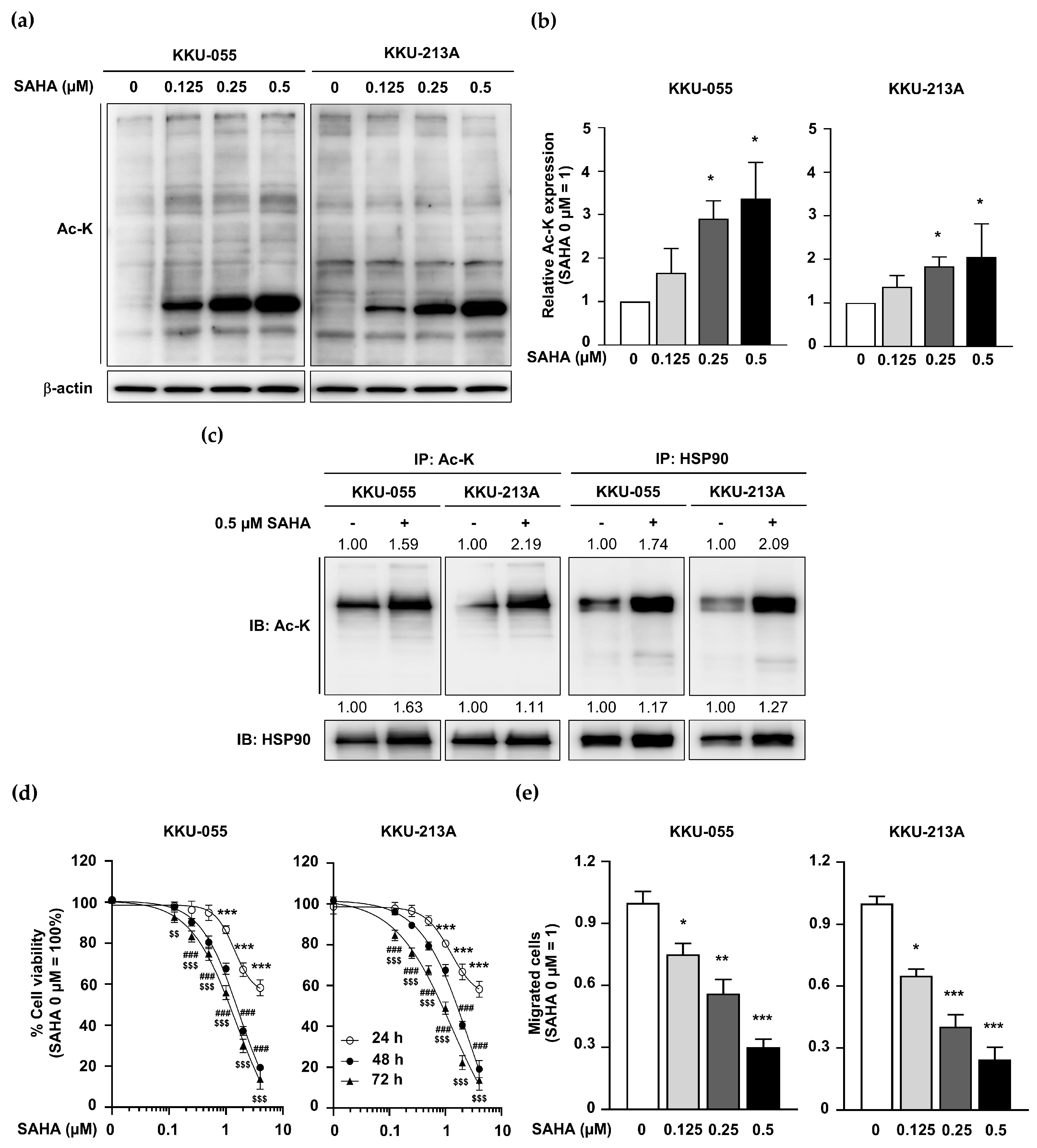
2.5. KDAC Inhibitor SAHA Induced HSP90 Acetylation but Inhibited CCA Cell Proliferation and Migration
2.6. ACC1 Inhibitory Conditions and SAHA Treatment Diminished GSK3β/Snail Signaling in an AKT-Dependent Manner
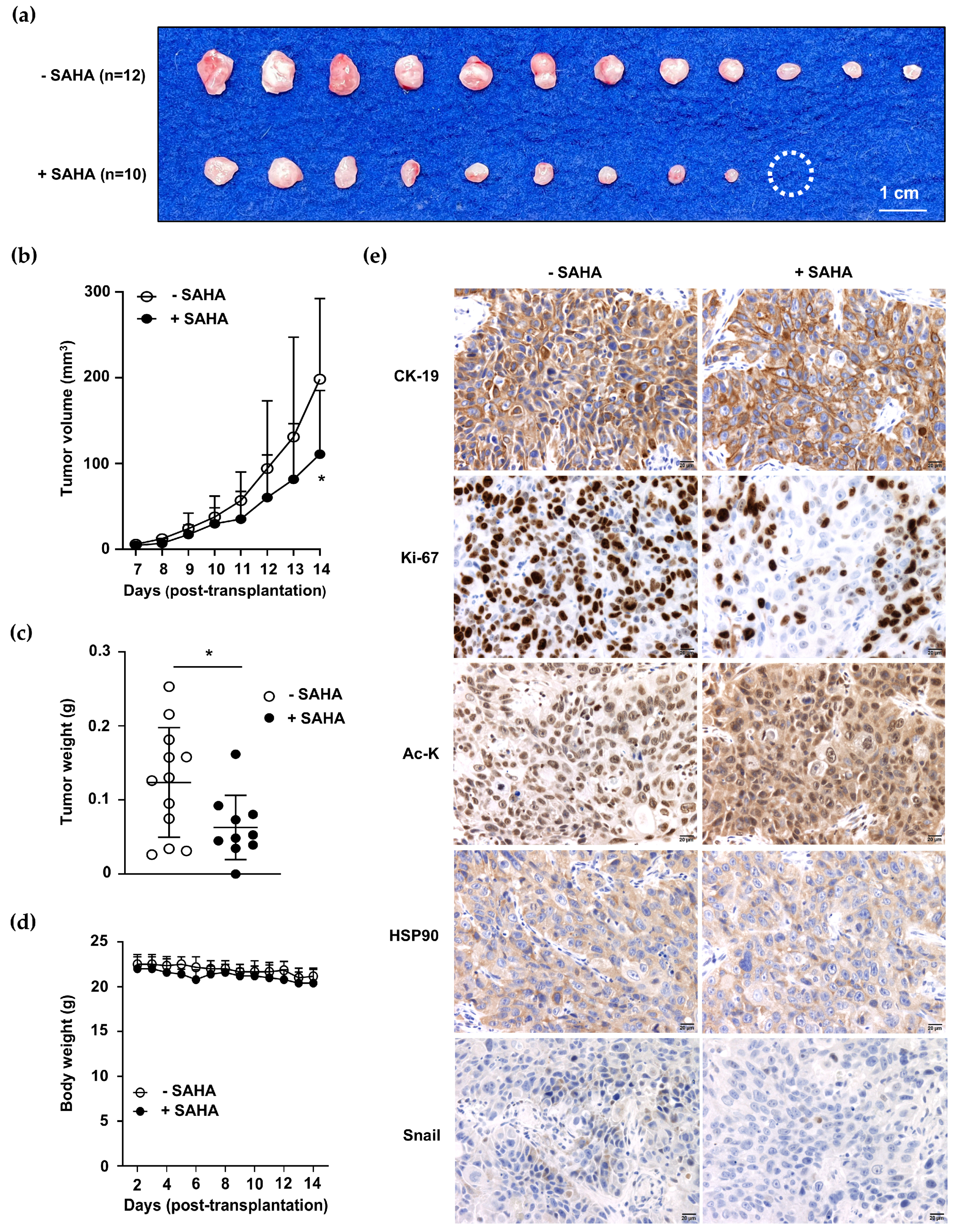
2.7. Decreased Tumor Growth Was Observed in Acquired Hyperacetylated ACC1-Deficient CCA Cells
2.8. SAHA Treatment Promoted Global Protein Acetylation but Inhibited CCA Growth
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Antibodies and Reagents
4.3. Molecular Docking Analysis
4.4. Clonogenic Assay
4.5. Cell Migration Assay
4.6. MTT Assay
4.7. Protein Extraction and Western Blot Analysis
4.8. Immunoprecipitation
4.9. Identification of Acetylated Peptides Using In-Gel Digestion, Liquid Chromatography–Tandem Mass Spectrometry Analysis
4.10. Animal Experiments
4.11. Immunohistochemistry Staining
4.12. Gene Expression Profiling from Databases
4.13. Statistical Analysis and Illustration Creation
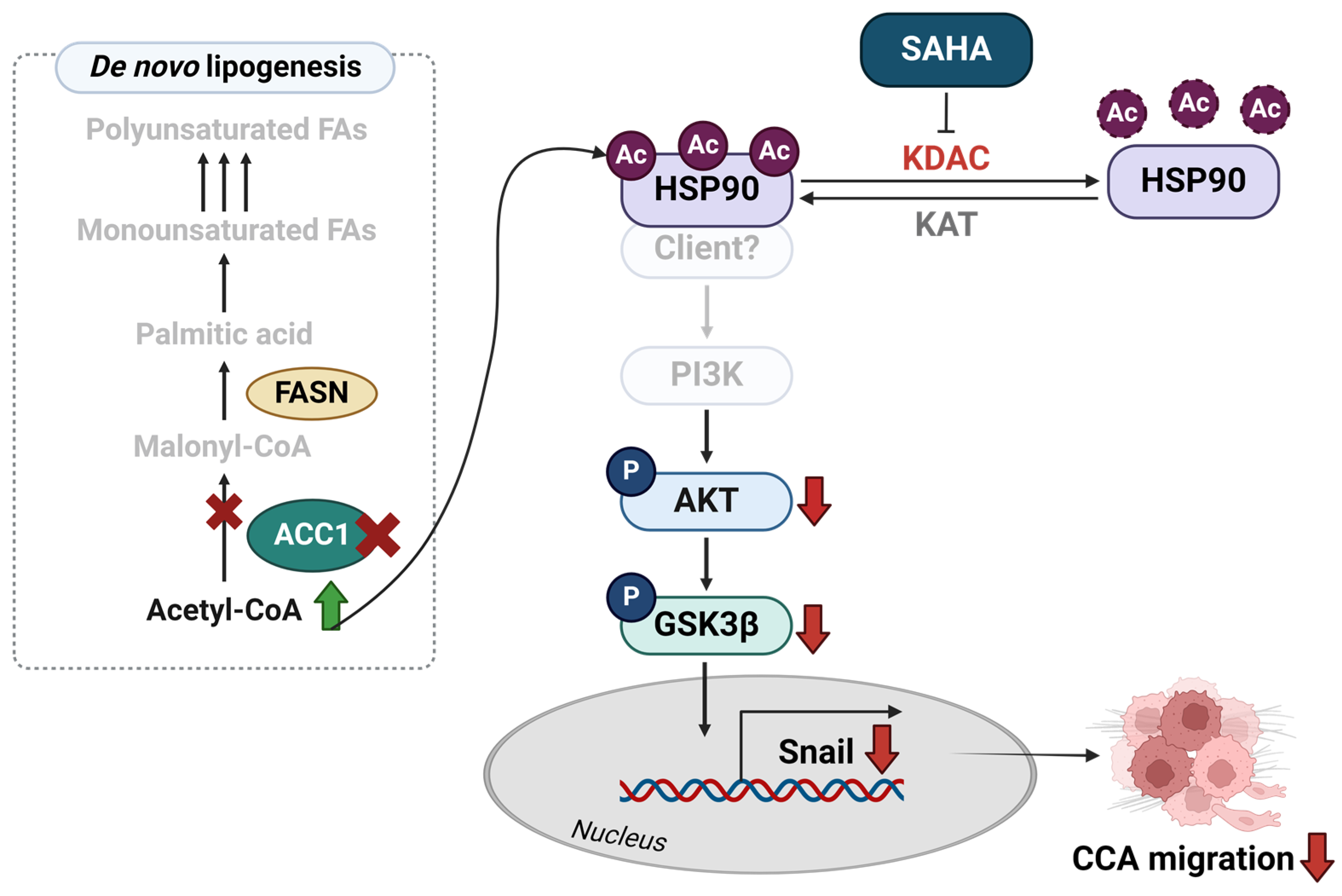
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Sripa, B.; Pairojkul, C. Cholangiocarcinoma: Lessons from Thailand. Curr. Opin. Gastroenterol. 2008, 24, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Kamel, I.; Cosgrove, D.P.; Herman, J.M.; Pawlik, T.M. Hilar cholangiocarcinoma: Diagnosis, treatment options, and management. Hepatobiliary Surg. Nutr. 2014, 3, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Phoomak, C.; Vaeteewoottacharn, K.; Silsirivanit, A.; Saengboonmee, C.; Seubwai, W.; Sawanyawisuth, K.; Wongkham, C.; Wongkham, S. High glucose levels boost the aggressiveness of highly metastatic cholangiocarcinoma cells via O-GlcNAcylation. Sci. Rep. 2017, 7, 43842. [Google Scholar] [CrossRef] [PubMed]
- Saisomboon, S.; Kariya, R.; Boonnate, P.; Sawanyawisuth, K.; Cháon, U.; Luvira, V.; Chamgramol, Y.; Pairojkul, C.; Seubwai, W.; Silsirivanit, A.; et al. Diminishing acetyl-CoA carboxylase 1 attenuates CCA migration via AMPK-NF-kappaB-snail axis. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166694. [Google Scholar] [CrossRef]
- Tong, L. Structure and function of biotin-dependent carboxylases. Cell Mol. Life Sci. 2013, 70, 863–891. [Google Scholar] [CrossRef]
- Boonnate, P.; Kariya, R.; Saranaruk, P.; Cha’on, U.; Sawanyawisuth, K.; Jitrapakdee, S.; Okada, S.; Vaeteewoottacharn, K. Five-(Tetradecyloxy)-2-furoic Acid Alleviates Cholangiocarcinoma Growth by Inhibition of Cell-cycle Progression and Induction of Apoptosis. Anticancer Res. 2021, 41, 3389–3400. [Google Scholar] [CrossRef]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Muller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855 e845. [Google Scholar] [CrossRef]
- Zaib, S.; Rana, N.; Khan, I. Histone Modifications and their Role in Epigenetics of Cancer. Curr. Med. Chem. 2022, 29, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Peela, N.; Barrientos, E.S.; Truong, D.; Mouneimne, G.; Nikkhah, M. Effect of suberoylanilide hydroxamic acid (SAHA) on breast cancer cells within a tumor-stroma microfluidic model. Integr. Biol. 2017, 9, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Fu, Y.; Zhang, Y.; Peng, F.; Lu, M.; Feng, Y.; Chen, L.; Chen, Z.; Li, M.; Chen, Y. Dissection of Anti-tumor Activity of Histone Deacetylase Inhibitor SAHA in Nasopharyngeal Carcinoma Cells via Quantitative Phosphoproteomics. Front. Cell Dev. Biol. 2020, 8, 577784. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhang, M.; Dorfman, R.G.; Li, Y.; Zhao, Z.; Pan, Y.; Zhou, Q.; Huang, S.; Zhao, S.; Yao, Y.; et al. Histone deacetylase 3 overexpression in human cholangiocarcinoma and promotion of cell growth via apoptosis inhibition. Cell Death Dis. 2017, 8, e2856. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.E.; Park, S.B.; Kim, K.; Kim, C.; Song, S.Y. CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci. Rep. 2017, 7, 10921. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, W.; Li, S.; Guo, D.; He, J.; Wang, Y. Acetyl-CoA Carboxylases and Diseases. Front. Oncol. 2022, 12, 836058. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Sun, L.; Wan, J.; Xu, R.; He, S.; Zhu, X. Tensin4 promotes invasion and migration of gastric cancer cells via regulating AKT/GSK-3beta/snail signaling pathway. Pathol. Res. Pract. 2020, 216, 153001. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef]
- Dana, P.; Saisomboon, S.; Kariya, R.; Okada, S.; Obchoei, S.; Sawanyawisuth, K.; Wongkham, C.; Pairojkul, C.; Wongkham, S.; Vaeteewoottacharn, K. CD147 augmented monocarboxylate transporter-1/4 expression through modulation of the Akt-FoxO3-NF-kappaB pathway promotes cholangiocarcinoma migration and invasion. Cell Oncol. 2020, 43, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Dokduang, H.; Jamnongkarn, W.; Promraksa, B.; Suksawat, M.; Padthaisong, S.; Thanee, M.; Phetcharaburanin, J.; Namwat, N.; Sangkhamanon, S.; Titapun, A.; et al. In vitro and in vivo Anti-Tumor Effects of Pan-HER Inhibitor Varlitinib on Cholangiocarcinoma Cell Lines. Drug Des. Devel Ther. 2020, 14, 2319–2334. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef]
- Fiskus, W.; Ren, Y.; Mohapatra, A.; Bali, P.; Mandawat, A.; Rao, R.; Herger, B.; Yang, Y.; Atadja, P.; Wu, J.; et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: A result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 2007, 13, 4882–4890. [Google Scholar] [CrossRef]
- Dey, G.; Bharti, R.; Dhanarajan, G.; Das, S.; Dey, K.K.; Kumar, B.N.; Sen, R.; Mandal, M. Marine lipopeptide Iturin A inhibits Akt mediated GSK3beta and FoxO3a signaling and triggers apoptosis in breast cancer. Sci. Rep. 2015, 5, 10316. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K.; Wang, Y. Posttranslational modification and beyond: Interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021, 27, 110. [Google Scholar] [CrossRef] [PubMed]
- Sripa, B.; Seubwai, W.; Vaeteewoottacharn, K.; Sawanyawisuth, K.; Silsirivanit, A.; Kaewkong, W.; Muisuk, K.; Dana, P.; Phoomak, C.; Lert-Itthiporn, W.; et al. Functional and genetic characterization of three cell lines derived from a single tumor of an Opisthorchis viverrini-associated cholangiocarcinoma patient. Hum. Cell 2020, 33, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [PubMed]
- Madauss, K.P.; Burkhart, W.A.; Consler, T.G.; Cowan, D.J.; Gottschalk, W.K.; Miller, A.B.; Short, S.A.; Tran, T.B.; Williams, S.P. The human ACC2 CT-domain C-terminus is required for full functionality and has a novel twist. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Koenig, T.; Menze, B.H.; Kirchner, M.; Monigatti, F.; Parker, K.C.; Patterson, T.; Steen, J.J.; Hamprecht, F.A.; Steen, H. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J. Proteome Res. 2008, 7, 3708–3717. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Ono, A.; Hattori, S.; Kariya, R.; Iwanaga, S.; Taura, M.; Harada, H.; Suzu, S.; Okada, S. Comparative study of human hematopoietic cell engraftment into BALB/c and C57BL/6 strain of rag-2/jak3 double-deficient mice. J. Biomed. Biotechnol. 2011, 2011, 539748. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Harlow, E.; Lane, D. Preparing paraffin tissue sections for immunostaining. CSH Protoc. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saisomboon, S.; Kariya, R.; Mahalapbutr, P.; Insawang, T.; Sawanyawisuth, K.; Cha’on, U.; Rungrotmongkol, T.; Wongkham, S.; Jitrapakdee, S.; Okada, S.; et al. Augmented Global Protein Acetylation Diminishes Cell Growth and Migration of Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2024, 25, 10170. https://doi.org/10.3390/ijms251810170
Saisomboon S, Kariya R, Mahalapbutr P, Insawang T, Sawanyawisuth K, Cha’on U, Rungrotmongkol T, Wongkham S, Jitrapakdee S, Okada S, et al. Augmented Global Protein Acetylation Diminishes Cell Growth and Migration of Cholangiocarcinoma Cells. International Journal of Molecular Sciences. 2024; 25(18):10170. https://doi.org/10.3390/ijms251810170
Chicago/Turabian StyleSaisomboon, Saowaluk, Ryusho Kariya, Panupong Mahalapbutr, Tonkla Insawang, Kanlayanee Sawanyawisuth, Ubon Cha’on, Thanyada Rungrotmongkol, Sopit Wongkham, Sarawut Jitrapakdee, Seiji Okada, and et al. 2024. "Augmented Global Protein Acetylation Diminishes Cell Growth and Migration of Cholangiocarcinoma Cells" International Journal of Molecular Sciences 25, no. 18: 10170. https://doi.org/10.3390/ijms251810170