
Chloroplast Genome Variation and Phylogenetic Relationships of Autochthonous Varieties of Vitis vinifera from the Don Valley


Abstract
:1. Introduction
2. Results
2.1. Subsection Characterization of the CP Genome Structure of Vitis vinifera Variety
2.2. IR Boundary Analysis
2.3. Genomic Sequence Divergence
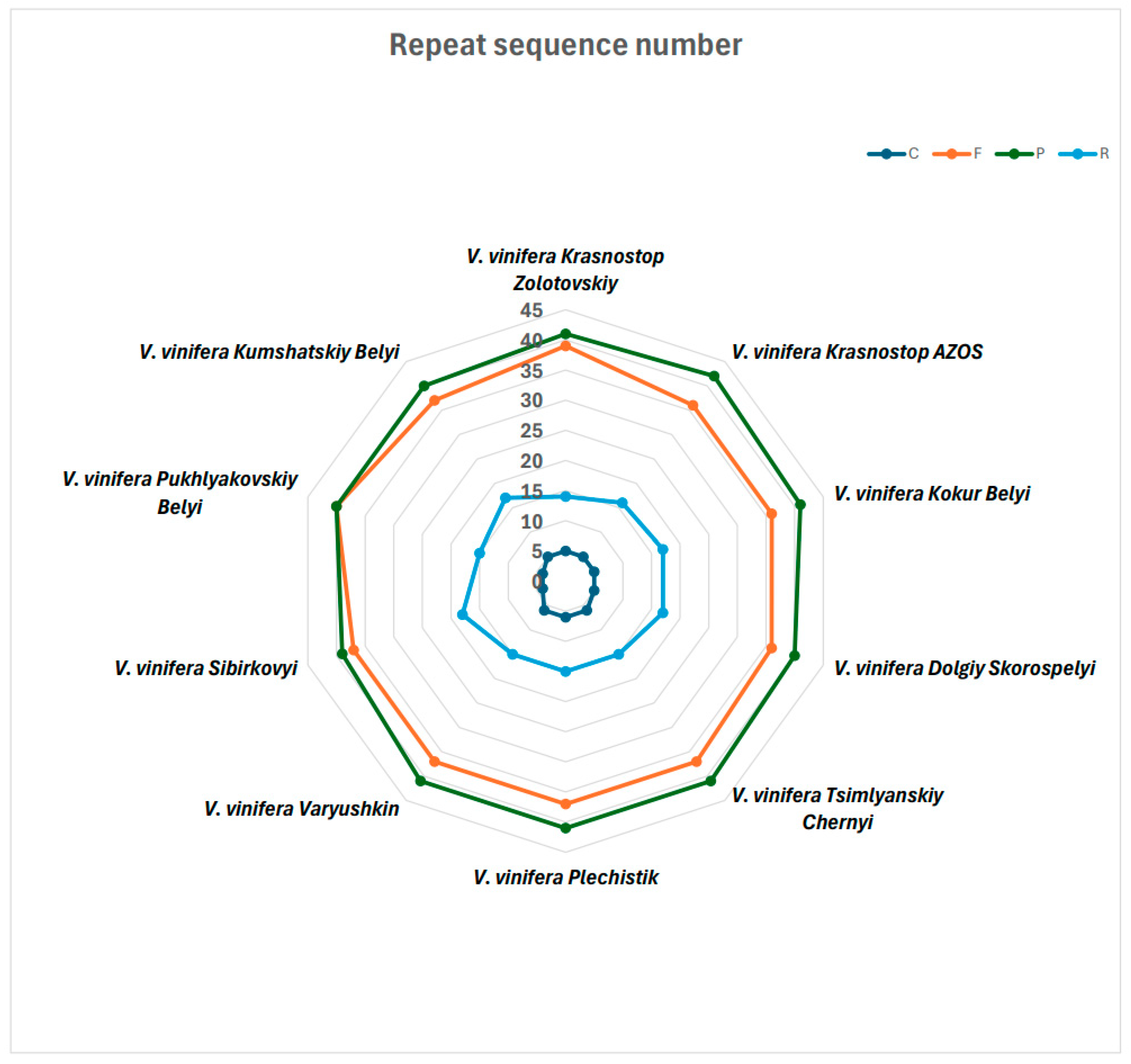
2.4. Identification of Long Repeats
2.5. Identification Simple Sequence Repeats
2.6. Selection Pressure Analysis
2.7. Codon Usage Bias
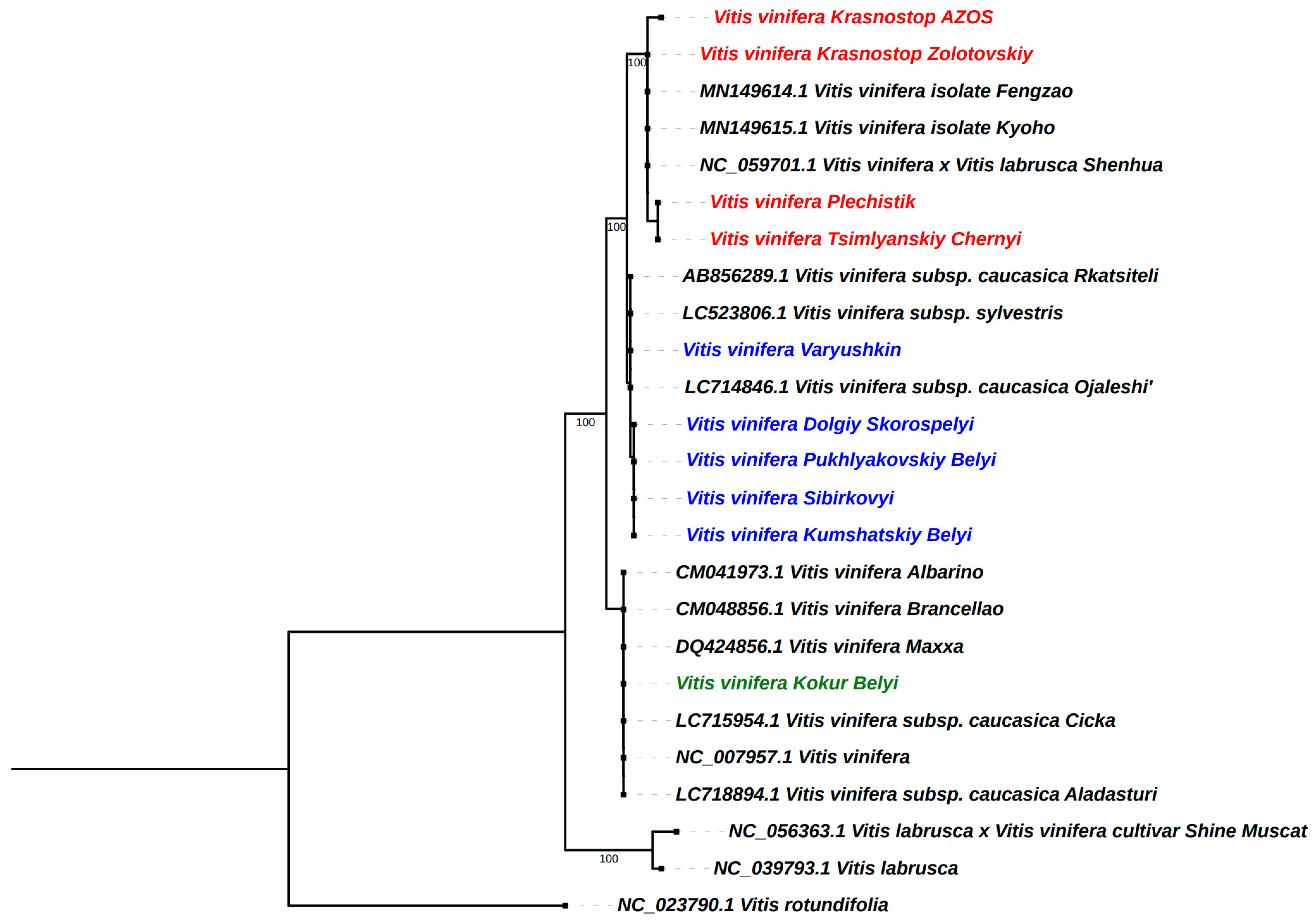
2.8. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials, DNA Extraction, and Sequencing
4.2. Genome Assembly and Annotation
4.3. Comparative Genome Analysis
4.4. Analysis of the Cp Genome for Repetitive Sequences
4.5. Codon Preference Analysis
4.6. Selective Analysis
4.7. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petrova, K.O.; Fedosov, D.Y.; Korzhenkov, A.A.; Toshchakov, S.V.; Patrushev, M.V.; Chernikovich, V.Y. On the gene pool of don valley grape varieties. Nanobiotechnol. Rep. 2022, 17, 722–730. [Google Scholar] [CrossRef]
- Mihaljević, M.Ž.; Maletić, E.; Preiner, D.; Zdunić, G.; Bubola, M.; Zyprian, E.; Pejić, I. Genetic diversity, population structure, and parentage analysis of Croatian grapevine germplasm. Genes 2020, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Manatskov, A.G.; Petrov, V.S.; Naumova, L.G. Autochthonous grape varieties in the Lower Don Region. Sadovod. Vinograd. 2024, 2, 22–29. [Google Scholar] [CrossRef]
- Kraysvetniy, M.I. The nart epos and the don winemaking. Russ. Vine 2018, 8, 116–131. [Google Scholar] [CrossRef]
- Anderson, K. JANCIS ROBINSON, JULIA HARDING and JOSÉ VOUILLAMOZ: Wine Grapes: A Complete Guide to 1368 Vine Varieties, including their Origins and Flavours. Ecco (Harper Collins), New York, October 2012, xxxvii + 1242 pp., ISBN 978-0062206367 (hardback), US$175. J. Wine Econ. 2013, 8, 106–109. [Google Scholar] [CrossRef]
- Pankin, M.I.; Petrov, V.S.; Lukianova, A.A.; Ilnitskaya, E.T.; Nikulushkina, G.E.; Kovalenko, A.G.; Bolshakov, V.A. The anapa ampelographic collection is the largest center of vine gene pool accumulation and research in Russia. Vavilov J. Genet. Breed. 2018, 22, 54–59. [Google Scholar] [CrossRef]
- Rybalko, E.; Ostroukhova, E.; Peskova, I.; Romanov, A.; Boyko, V. Crimean autochthonous grape varieties as a factor of high-quality winemaking in a changing climate. BIO Web Conf. 2022, 53, 01001. [Google Scholar] [CrossRef]
- Volynkin, V.; Polulyakh, A.; Levchenko, S.; Vasylyk, I.; Likhovskoy, V. Aspects of the particular genetics of grapes prolonged for all horticulture crops. In Horticultural Crops; Kossi Baimey, H., Hamamouch, N., Adjiguita Kolombia, Y., Eds.; IntechOpen: London, UK, 2020; ISBN 978-1-83880-421-3. [Google Scholar]
- Il’nitskaya, E.T.; Tokmakov, S.V.; Suprun, I.I.; Naumova, L.G.; Ganich, V.A. Genetic similarity of the autochthonous grapevine varieties from don region revealed by SSR-analysis and main leaf ampelographic traits. Sel’skokhozyaistvennaya Biol. 2016, 51, 60–67. [Google Scholar] [CrossRef]
- Ganich, V.A.; Naumova, L.G. Kumshatsky Belyy—Perspective aborigenous Don grapevine variety. Bull. KrasGAU 2021, 12, 11–16. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Robbins, E.H.J.; Kelly, S. The evolutionary constraints on angiosperm chloroplast adaptation. Genome Biol. Evol. 2023, 15, evad101. [Google Scholar] [CrossRef] [PubMed]
- Sharko, F.; Gladysheva-Azgari, M.; Tsygankova, S.; Mitrofanova, I.; Boulygina, E.; Slobodova, N.; Smykov, A.; Rastorguev, S.; Nedoluzhko, A. The complete chloroplast genome sequence of cultivated Prunus persica cv. “Sovetskiy”. Mitochondrial DNA Part B Resour. 2021, 6, 2882–2883. [Google Scholar] [CrossRef]
- Gao, H.; McCormick, A.J.; Roston, R.L.; Lu, Y. Editorial: Structure and function of chloroplasts, Volume III. Front. Plant Sci. 2023, 14, 1145680. [Google Scholar] [CrossRef] [PubMed]
- Bock, R. Structure, function, and inheritance of plastid genomes. In Cell and Molecular Biology of Plastids; Bock, R., Ed.; Topics in Current Genetics; Springer: Berlin/Heidelberg, Germany, 2007; Volume 19, pp. 29–63. ISBN 978-3-540-75375-9. [Google Scholar]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Umesono, K.; Shiki, Y.; Takeuchi, M.; Chang, Z.; et al. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 1986, 322, 572–574. [Google Scholar] [CrossRef]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Advances in Photosynthesis and Respiration; Springer: Dordrecht, The Netherlands, 2012; Volume 35, pp. 103–126. ISBN 978-94-007-2919-3. [Google Scholar]
- Herrmann, L.; Haase, I.; Blauhut, M.; Barz, N.; Fischer, M. DNA-based differentiation of the Ecuadorian cocoa types CCN-51 and Arriba based on sequence differences in the chloroplast genome. J. Agric. Food Chem. 2014, 62, 12118–12127. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.L.; Asaf, S.; Lee, I.-J.; Al-Harrasi, A.; Al-Rawahi, A. First chloroplast genomics study of Phoenix dactylifera (var. Naghal and Khanezi): A comparative analysis. PLoS ONE 2018, 13, e0200104. [Google Scholar] [CrossRef]
- Kahlau, S.; Aspinall, S.; Gray, J.C.; Bock, R. Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J. Mol. Evol. 2006, 63, 194–207. [Google Scholar] [CrossRef]
- Tang, J.; Xia, H.; Cao, M.; Zhang, X.; Zeng, W.; Hu, S.; Tong, W.; Wang, J.; Wang, J.; Yu, J.; et al. A comparison of rice chloroplast genomes. Plant Physiol. 2004, 135, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, R.; Cultrera, N.G.M.; Díez, C.M.; Baldoni, L.; Rubini, A. Identification of new polymorphic regions and differentiation of cultivated olives (Olea europaea L.) through plastome sequence comparison. BMC Plant Biol. 2010, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Li, Q. The Complete Chloroplast Genome of Endangered Species Stemona parviflora: Insight into the Phylogenetic Relationship and Conservation Implications. Genes 2022, 13, 1361. [Google Scholar] [CrossRef]
- Bock, R. Engineering plastid genomes: Methods, tools, and applications in basic research and biotechnology. Annu. Rev. Plant Biol. 2015, 66, 211–241. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 2020, 53, 21. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Li, Z.-X.; Xie, D.-F.; Gui, L.-J.; Peng, C.; Wen, J.; He, X.-J. Plastomes of eight Ligusticum species: Characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol. 2020, 20, 519. [Google Scholar] [CrossRef]
- Gladysheva-Azgari, M.; Sharko, F.; Slobodova, N.; Petrova, K.; Boulygina, E.; Tsygankova, S.; Mitrofanova, I. Comparative Analysis Revealed Intrageneric and Intraspecific Genomic Variation in Chloroplast Genomes of Actinidia spp. (Actinidiaceae, Viridiplantae). Horticulturae 2023, 9, 1175. [Google Scholar] [CrossRef]
- Nie, L.; Cui, Y.; Wu, L.; Zhou, J.; Xu, Z.; Li, Y.; Li, X.; Wang, Y.; Yao, H. Gene Losses and Variations in Chloroplast Genome of Parasitic Plant Macrosolen and Phylogenetic Relationships within Santalales. Int. J. Mol. Sci. 2019, 20, 5812. [Google Scholar] [CrossRef]
- Sharko, F.S.; Petrova, K.O.; Molchanov, N.V.; Patrushev, M.V.; Fedosov, D.Y.; Toshchakov, S.V. The complete chloroplast genome sequence of Vitis vinifera ‘Krasnostop Zolotovskiy’, an autochthonous variety of the Don Valley. Turczaninowia 2024, 27, 124–129. [Google Scholar] [CrossRef]
- Fedosov, D.Y.; Korzhenkov, A.A.; Petrova, K.O.; Sapsay, A.O.; Sharko, F.S.; Toshchakov, S.V.; Kolosova, A.A.; Bakhmutova, E.D.; Patrushev, M.V. SNP-Based Analysis Reveals Authenticity and Genetic Similarity of Russian Indigenous V. vinifera Grape Cultivars. Plants 2021, 10, 2696. [Google Scholar] [CrossRef] [PubMed]
- Meger, J.; Ulaszewski, B.; Vendramin, G.G.; Burczyk, J. Using reduced representation libraries sequencing methods to identify cpDNA polymorphisms in European beech (Fagus sylvatica L.). Tree Genet. Genomes 2019, 15, 7. [Google Scholar] [CrossRef]
- Somaratne, Y.; Guan, D.-L.; Wang, W.-Q.; Zhao, L.; Xu, S.-Q. The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 2019, 9, 51. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Jarvis, P.; López-Juez, E. Biogenesis and homeostasis of chloroplasts and other plastids. Nat. Rev. Mol. Cell Biol. 2013, 14, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- George, B.; Bhatt, B.S.; Awasthi, M.; George, B.; Singh, A.K. Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 2015, 61, 665–677. [Google Scholar] [CrossRef]
- Qin, Z.; Cai, Z.; Xia, G.; Wang, M. Synonymous codon usage bias is correlative to intron number and shows disequilibrium among exons in plants. BMC Genom. 2013, 14, 56. [Google Scholar] [CrossRef]
- Jiao, L.; Lu, Y.; He, T.; Li, J.; Yin, Y. A strategy for developing high-resolution DNA barcodes for species discrimination of wood specimens using the complete chloroplast genome of three Pterocarpus species. Planta 2019, 250, 95–104. [Google Scholar] [CrossRef]
- Lo Piccolo, S.; Alfonzo, G.; Conigliaro, G.; Moschetti, G.; Burruano, S.; Barone, A. A simple and rapid DNA extraction method from leaves of grapevine suitable for polymerase chain reaction analysis. Afr. J. Biotechnol. 2012, 11, 10305. [Google Scholar] [CrossRef]
- Gladysheva-Azgari, M.; Petrova, K.; Tsygankova, S.; Mitrofanova, I.; Smykov, A.; Boulygina, E.; Slobodova, N.; Rastorguev, S.; Sharko, F. A de novo genome assembly of cultivated Prunus persica cv. “Sovetskiy”. PLoS ONE 2022, 17, e0269284. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; NISC Comparative Sequencing Program; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. Dnasp 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, bbx108. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index–A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Using RAxML to infer phylogenies. Curr. Protoc. Bioinform. 2015, 51, 6.14.1–6.14.14. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group | Genes |
|---|---|---|
| Photosynthesis | Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ, psbN | |
| ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Cytochrome b/f complex | petA, petB *, petD *, petN, petL, petG | |
| Cytochrome C synthesis | ccsA | |
| Rubisco | rbcL | |
| NADPH dehydrogenase | ndhA *, ndhB (×2) *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Self-replication | RNA polymerase | rpoA, rpoB, rpoC2, rpoC1 * |
| Ribosomal proteins | rps2 (×2), rps3, rps4, rps7 (×2), rps8, rps11, rps12 (×2) *, rps14, rps15, rps16 *, rps18, rps19, rpl2 *, rpl14, rpl16 *, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 | |
| Translation initiation factor | infA | |
| Ribosomal RNA | rrn16 (×2), rrn23 (×2), | |
| rrn4.5 (×2), rrn5 (×2) | ||
| Transfer RNA | trna-UGC (×2) *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-CAU (×2), trnI-GAU * (×2), trnK-UUU *, trnL-CAA (×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnV-UAC *, trnW-CCA, trnY-GUA | |
| Other genes | Maturase | matK |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| Proteolysis | ClpP ** | |
| Conserved ORFs | ycf1 (×2), ycf2 (×2), ycf3, ycf4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharko, F.S.; Petrova, K.O.; Patrushev, M.V.; Fedosov, D.Y.; Toshchakov, S.V. Chloroplast Genome Variation and Phylogenetic Relationships of Autochthonous Varieties of Vitis vinifera from the Don Valley. Int. J. Mol. Sci. 2024, 25, 9928. https://doi.org/10.3390/ijms25189928
Sharko FS, Petrova KO, Patrushev MV, Fedosov DY, Toshchakov SV. Chloroplast Genome Variation and Phylogenetic Relationships of Autochthonous Varieties of Vitis vinifera from the Don Valley. International Journal of Molecular Sciences. 2024; 25(18):9928. https://doi.org/10.3390/ijms25189928
Chicago/Turabian StyleSharko, F. S., K. O. Petrova, M. V. Patrushev, D. Y. Fedosov, and S. V. Toshchakov. 2024. "Chloroplast Genome Variation and Phylogenetic Relationships of Autochthonous Varieties of Vitis vinifera from the Don Valley" International Journal of Molecular Sciences 25, no. 18: 9928. https://doi.org/10.3390/ijms25189928